
Abstract
Five BCR-ABL1 tyrosine kinase inhibitors (TKIs), imatinib, nilotinib, dasatinib, bosutinib, and ponatinib, are currently approved for the treatment of chronic myeloid leukemia (CML). Standard treatment of CML with TKIs is highly effective in reducing disease burden, delaying disease progression, and prolonging overall survival of patients; however, resistance to TKI treatment has become an increasingly important cause of treatment failure. The emergence of mutations in the BCR-ABL1 kinase domain is a common mechanism of TKI resistance, and laboratory testing to detect these mutations is currently available for clinical use. Patients who do not respond or have lost their response to TKI therapy should be considered for mutational testing. Despite clinical practice guidelines that recommend testing for BCR-ABL1 mutations in patients with clinical signs of TKI resistance, many oncologists and hematologists managing patients with CML do not perform such testing. This review addresses outstanding questions related to when testing should be conducted, what type of testing should be done, and how testing results should be applied to subsequent therapeutic decisions. It describes how BCR-ABL1 kinase domain mutations confer resistance, outlines the prevalence of mutations in patients with resistance to TKIs, summarizes the common and investigational methods used in mutational testing, and presents an algorithm reflecting a clinical perspective on how and when to conduct mutational testing, and what to do with test results.
Introduction
The introduction of tyrosine kinase inhibitors (TKIs) against the aberrant BCR-ABL1 kinase has dramatically improved the clinical outlook of patients with chronic myeloid leukemia (CML). Five- and 8-year overall survival (OS) for patients diagnosed since imatinib (Gleevec; Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA) was first approved for CML are significantly higher than for patients from any time period before [Bjorkholm et al. 2011; Kantarjian et al. 2012a; Brunner et al. 2013]. Although TKI therapy can elicit high rates of treatment response in the majority of patients, some patients will develop resistance to TKI therapy. In the pivotal phase III International Randomized Study of Interferon and STI571 (IRIS) in newly diagnosed patients with CML in chronic phase (CP), rates of complete hematologic response (CHR; 95.3% versus 55.5%, p < 0.001) and complete cytogenetic response (CCyR; 73.8% versus 8.5%, p < 0.001) at 12 months were significantly higher with imatinib than with interferon α plus cytarabine [O’Brien et al. 2003]. Yet, these data imply that the patients in the imatinib arm who did not achieve CHR (4.7%) or CCyR (26.2%) at 12 months may have been intrinsically resistant to imatinib treatment. After 8 years of follow up on the IRIS study, 16% of patients in the imatinib arm had discontinued treatment because of unsatisfactory therapeutic outcome [Deininger et al. 2009], suggesting that these patients may have acquired resistance to imatinib over time.
Patients with resistance to first-line TKI treatment clearly require closer evaluation and follow up. However, while the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines) for CML provide treatment options based on BCR-ABL1 kinase domain mutation status, they make only general, not specific, recommendations for the evaluation of patients who demonstrate resistance to TKI therapy [NCCN, 2014]. In particular, NCCN Guidelines recommend BCR-ABL1 kinase domain mutation analysis but do not specify what type of testing should be done or how clinicians should weigh these test results when making subsequent therapeutic decisions. As a consequence of these gaps in guidance, when faced with a patient with suspected treatment resistance, only 58% of clinicians surveyed would order mutational analysis [Kantarjian et al. 2013].
This review describes how BCR-ABL1 kinase domain mutations confer resistance, outlines the prevalence of mutations in patients with resistance to TKIs, summarizes the common and investigational methods used in mutational analysis, and provides a clinical perspective on how and when to conduct mutational analysis, and what to do with test results.
BCR-ABL1 kinase domain mutations as a mechanism of resistance to TKIs
The ABL1 kinase comprises the N-terminal and C-terminal lobes [Reddy and Aggarwal, 2012]. The binding of adenosine triphosphate (ATP) to its binding site on ABL1, a deep pocket between the N- and C lobes, activates BCR-ABL1 tyrosine kinase activity. The P-loop, encompassing amino acid residues 244–255 of the ABL1 kinase domain, binds to the phosphate groups of ATP. ABL1 also includes an activation region, A-loop, at residues 381–402 that is responsible for regulating kinase activity [Deininger et al. 2005; Quintas-Cardama and Cortes, 2009; Reddy et al. 2012]. The A-loop includes a conserved DFG (aspartate–phenylalanine–glycine) motif in which the aspartate, and consequently the DFG motif, is pointed in towards the ATP-binding site (DFG-in) when the loop is in its open or active state, and out or away from the binding site in the closed or inactive conformation (DFG-out) [Deininger et al. 2005; Reddy et al. 2012]. A catalytic (C) domain at residues 350–363 is involved in key roles that are essential for catalytic reactions [Quintas-Cardama and Cortes, 2009; Reddy et al. 2012]. Mutations in these key regions of the ABL1 kinase domain can result in resistance to BCR-ABL1 TKIs. In addition, a ‘gatekeeper’ in the ATP binding pocket at residue 315, normally occupied by threonine, is a key binding site for a few BCR-ABL1 TKIs [Gorre et al. 2001; Reddy et al. 2012]. Substitution of threonine for isoleucine (T315I) deters binding of some BCR-ABL1 TKIs to the ABL1 kinase by eliminating the potential for hydrogen bonding or creating steric hindrance [Gorre et al. 2001; Bixby and Talpaz, 2009; Quintas-Cardama and Cortes, 2009].
The chemical structure of each of the five TKIs approved for treatment of CML is shown in Figure 1 and important contacts between each TKI and the ABL1 kinase domain are shown in Figure 2. Although all the TKIs act as competitive inhibitors of ATP binding to the ABL1 kinase, the chemical structural differences between the TKIs underlie the distinct molecular interactions between each TKI and the ATP-binding site of ABL1 kinase. As a result, the TKIs are susceptible to an overlapping, yet distinct, array of kinase domain mutations.

Chemical structures of the BCR-ABL1 tyrosine kinase inhibitors approved for treatment of chronic myeloid leukemia (CML). Notable chemical moieties are indicated: ethinyl (yellow); methylimidazole (light green); phenyl-amino group (tan); piperazine (red); pyridine (gray); pyrimidine (blue); quinolinecarbonitrile group (turquoise); thiazole carboxamide group (orange); trifluoromethyl group (purple).

Hydrogen bond interactions (red double-headed arrows) between atoms of the tyrosine kinase inhibitors and amino acids of the ABL1 kinase (red font).
In theoretical terms, any kinase domain mutation that causes steric hindrance blocking TKI binding or that stabilizes the active conformation of the ABL1 kinase can result in TKI resistance. If, however, a mutation produces a protein configuration so deleterious that kinase activity is destroyed altogether, then the mutation would not be propagated [O’Hare et al. 2007]. Therefore, only mutations that affect TKI binding without drastically affecting kinase activity are capable of conferring resistance. As an illustration of this point, mutations that might change the methionine at position 318 of the BCR-ABL1 kinase are not detected in patients, despite the importance of this residue in making hydrogen bond interactions with each of the TKIs.
Imatinib
Imatinib binds to the ATP-binding site within the catalytic site of ABL1 and stabilizes the inactive form of the kinase, with the A-loop in a closed DFG-out conformation and the ATP-binding P-loop in a distorted conformation [Schindler et al. 2000].
Nilotinib
Nilotinib was designed to be more potent and selective than imatinib. Like imatinib, nilotinib binds to the inactive form of the ABL1 kinase [Weisberg et al. 2005], and it is not surprising that many kinase domain mutants are insensitive to both imatinib and nilotinib [O’Hare et al. 2007]. Unlike imatinib, however, nilotinib has a methylimidazole ring that fits in a deeper hydrophobic pocket that is created when the ABL1 kinase is in the inactive, DFG-out conformation [Weisberg et al. 2005; Manley et al. 2010]. The importance of the hydrophobic pocket to the mechanism of action of nilotinib suggests that mutations altering the hydrophobicity of the pocket might be expected to confer considerable resistance to nilotinib. There are, in fact, only a limited number of observed mutations that affect nilotinib sensitivity but not imatinib sensitivity [O’Hare et al. 2007], consistent with the theory that mutations affecting the hydrophobicity of the pocket are deleterious to the kinase. Indeed, most mutations that confer in vitro insensitivity to nilotinib primarily affect the A-loop, the P-loop, and the T315 residue of the ATP-binding site [O’Hare et al. 2005; Redaelli et al. 2009].
Dasatinib
Dasatinib is a thiazole carboxamide that inhibits both the ABL1 and the SRC kinases [Lombardo et al. 2004]. Unlike imatinib and nilotinib, dasatinib binds the open, DFG-in conformation of the ABL1 kinase [Lombardo et al. 2004]. Because dasatinib exerts its inhibitory action via the ABL1 ATP-binding site, mutations in the ATP-binding region and the P-loop decrease in vitro sensitivity of the ABL1 kinase to dasatinib. Relative to imatinib and nilotinib, however, dasatinib is less affected by mutations in the A-loop [Redaelli et al. 2009].
Bosutinib
Bosutinib, like dasatinib, is a dual SRC/ABL1 inhibitor [Golas et al. 2003]. Either conformation of the DFG triad, in or out, can be accommodated by bosutinib [Levinson and Boxer, 2012], making it theoretically possible for bosutinib to bind both the active and inactive forms of the ABL1 kinase. Bosutinib makes a hydrogen bond with only one amino acid, M318, and makes only van der Waals contacts, not hydrogen bond interactions, with T315 [Levinson and Boxer, 2012]. Nevertheless, because T315 makes van der Waals contacts with two chemical groups on bosutinib, this TKI is insensitive to mutations affecting T315 [Levinson and Boxer, 2012]. The P-loop of the ABL1 kinase makes no contacts with bosutinib [Levinson and Boxer, 2012], a property that is reflected by in vitro data showing that bosutinib retains a degree of sensitivity to P-loop mutations [Redaelli et al. 2009].
Ponatinib
Ponatinib is a BCR-ABL1 inhibitor that binds the closed, DFG-out conformation of the ABL1 kinase domain [Zhou et al. 2011]. A unique property of ponatinib is its lack of hydrogen bond formation with T315 [Zhou et al. 2011]. When ponatinib is complexed with the T315I mutant ABL1 kinase, its binding to the kinase is not sterically hindered by the isoleucine at position 315. That is because the ethinyl moiety of ponatinib allows the TKI to be displaced slightly from the ATP-binding site to form favorable carbon–carbon contacts with I315 [Zhou et al. 2011].
Ponatinib makes close contact with the P-loop and forms a hydrogen bond with F317. Mutations that destabilize the P-loop (e.g., mutations in E255) or disrupt interactions between ponatinib and F317 negatively affect ponatinib sensitivity in vitro [Zhou et al. 2011]. Although ponatinib makes weak van der Waals contacts with F359, this residue is involved in the formation of the hydrophobic pocket, and mutations that alter the conformation of this pocket could disrupt a key hydrogen bond interaction between ponatinib and E286 [Zhou et al. 2011].
Prevalence of BCR-ABL1 kinase domain mutations in patients with CML
In the pre-TKI era
Before the approval of BCR-ABL1 TKIs for the treatment of CML, standard therapy for CML-CP included interferon α, with or without cytarabine, and allogeneic stem cell transplantation [Faderl et al. 1999]. Other approved agents in common use were hydroxyurea and busulfan [Faderl et al. 1999]. Because the mechanisms of action of these medications do not directly impinge on the BCR-ABL1 kinase, resistance to these agents does not generally involve BCR-ABL1 kinase domain mutations [Razga et al. 2012]. Rather, mechanisms of resistance to these agents involve reduced drug influx into cells [Hait et al. 1993], activity of drug-inducible target proteins [Landolfo et al. 2000], and upregulation of gene mRNA and protein expression [Choy et al. 1988].
At baseline, prior to TKI therapy
The existence of TKI-resistant BCR-ABL1 kinase domain mutations prior to exposure to TKI therapy has been documented. One study found low levels of TKI-resistant mutant cells in 3 of 24 patients (13%) prior to treatment with imatinib. During treatment, the relative proportion of these mutant cells increased [Roche-Lestienne et al. 2002]. These findings suggest that TKI-resistant BCR-ABL1 mutations may be present in some patients with CML before treatment with TKIs, and that treatment with TKIs effectively selects for resistant clones by eliminating sensitive clones [Razga et al. 2012].
During first-line TKI therapy
Rates of resistance to TKIs noted in clinical studies depend in part on the specific TKI and on the definition of ‘resistance,’ which may differ, if provided at all, depending on the clinical study. Resistance was not defined in the IRIS study [O’Brien et al. 2003]. European LeukemiaNet (ELN) 2006 criteria [Baccarani et al. 2006] were used to define suboptimal response and treatment failure in Evaluating Nilotinib Efficacy and Safety in Clinical Trials, newly diagnosed patients (ENESTnd) [Saglio et al. 2010] and to define progression and treatment failure in Dasatinib Versus Imatinib Study In Treatment-Naive CML Patients (DASISION) [Kantarjian et al. 2010]. As a point of reference, if primary resistance were defined conservatively as the failure to achieve CCyR by 12 months and acquired resistance as disease progression, then the incidence of overall resistance in these pivotal phase III studies was 30–40% among imatinib-treated patients, and approximately 20% among nilotinib-treated and dasatinib-treated patients [O’Brien et al. 2003; Kantarjian et al. 2010; Saglio et al. 2010]. With resistance defined in this way, the incidence of TKI resistance appears to be lower with nilotinib and dasatinib than with imatinib.
The prevalence of BCR-ABL1 kinase domain mutations was not determined in any of these pivotal studies in the subgroup of patients with resistance. In ENESTnd, postbaseline mutational analysis was conducted in patients with lack of response, loss of response, or disease progression, and in patients who discontinued for any reason, including study completion (i.e. patients with and without clinical signs of resistance). In this broadly defined group of patients, BCR-ABL1 mutations were detected in 5% (n = 22/443) of patients on nilotinib and 9% (n = 21/237) of patients on imatinib [Hochhaus et al. 2013], indicating that the development of new BCR-ABL1 mutations is relatively infrequent among patients receiving TKI therapy. New BCR-ABL1 mutations on TKI therapy were nonetheless clinically significant: 14% (n = 3/22) and 33% (n = 7/21) of patients with new mutations on nilotinib and imatinib respectively progressed to accelerated phase (AP) or blast phase (BP) CML while on treatment in ENESTnd [Hochhaus et al. 2013]. For reference, after 3 years of follow up in the overall study population in ENESTnd, 0.9% (n = 5/563) on nilotinib and 4% (n = 12/283) on imatinib had progressed to AP/BP on treatment [Larson et al. 2012].
In DASISION, postbaseline mutational analysis was conducted only in patients who discontinued for any reason. In this narrowly defined group, BCR-ABL1 mutations were detected in 23% (n = 10/44) of patients on dasatinib and 20% (n = 10/49) of patients on imatinib. Of the patients with new mutations, 80% (n = 8/10) of patients on dasatinib and 70% (n = 7/10) of patients on imatinib experienced protocol-defined progression [doubling of white cell count to >20 × 109/L without CHR, loss of CHR, increase to >35% Philadelphia chromosome-positive (Ph+) metaphase cells, transformation to CML-AP/BP, or death from any cause] [Kantarjian et al. 2012b].
In studies that have assessed the prevalence of BCR-ABL1 mutations among patients who demonstrated clinical signs of resistance (e.g. lack of response, loss of response, or progression to advanced phases of CML on treatment), BCR-ABL1 kinase domain mutations were detected in 15–50% of patients on imatinib, 50% of patients on nilotinib, and 10–22% of patients on dasatinib [Lahaye et al. 2005; Jabbour et al. 2006; Soverini et al. 2006; Cortes et al. 2010; Kantarjian et al. 2012b; Radich et al. 2012], depending on the definition of resistance, the patient population, and the method of mutational testing used. These findings indicate that the development of TKI-resistant mutations is a common mechanism of resistance. Other mechanisms of resistance include BCR-ABL1-dependent mechanisms, such as BCR-ABL1 gene amplification, and BCR-ABL1-independent mechanisms, such as intracellular drug uptake and clonal evolution (reviewed by Quintas-Cardama and colleagues) [Quintas-Cardama et al. 2009]. An in-depth discussion of these mechanisms of resistance is beyond the scope of this review.
BCR-ABL1 kinase domain mutations detected on TKI therapy
Following first-line therapy
Specific BCR-ABL1 kinase domain mutations detected following first-line TKI therapy are summarized in Table 1. At present, information regarding mutations arising with first-line bosutinib and ponatinib treatment is limited, awaiting longer-term data from ongoing clinical studies.
BCR-ABL1 kinase domain mutations detected following TKI therapy in clinical studies.

Patients who had post-baseline BCR-ABL1 kinase domain mutational analysis done.
Of the patients who had mutational analysis done.
Total number of mutations detected may exceed the total number of patients with mutations because some patients may have more than one mutation.
54 of 198 (27%) patients with CP had mutations: 6 of 44 (14%) treated with imatinib frontline and 48 of 154 (31%) treated with imatinib after interferon α failure.
Of 443 patients who had mutational analysis done, 228 patients had received nilotinib 300 mg twice daily, and 215 patients had received nilotinib 400 mg twice daily.
–, not detected or not reported; ABR, adenosine triphosphate binding region; ALL, Philadelphia chromosome-positive acute lymphoblastic leukemia; AP, accelerated phase; BP, blast phase; CP, chronic phase; DASISION, Dasatinib Versus Imatinib Study In Treatment-Naive CML Patients; ENESTnd, Evaluating Nilotinib Efficacy and Safety in Clinical Trials–newly diagnosed patients; SBR, substrate-binding region; GIMEMA, Gruppo Italiano Malattie Ematologiche dell’ Adulto (Italian Group for Haematological Diseases in Adults).
With respect to the first-line treatment with imatinib, nilotinib, or dasatinib, mutations detected in patients in some instances are consistent with in vitro TKI sensitivity data, such as mutations in the P-loop and substrate-binding region in patients treated with imatinib or nilotinib, or mutations in the ATP-binding region in patients treated with dasatinib [O’Hare et al. 2007; Redaelli et al. 2009; Lombardo et al. 2004]. In other instances, however, in vivo mutations are not consistent with in vitro mutations. For example, mutations affecting F317 are infrequently detected in patients treated with imatinib or nilotinib, and mutations affecting the P-loop, substrate-binding region, and A-loop are infrequently detected in patients treated with dasatinib. Such inconsistency underscores an important point about the limits of correlating in vitro experimental results with what might occur in patients on therapy: the type of BCR-ABL1 mutation that might arise in patients treated with TKI therapy is not always predictable. Although mutations in the BCR-ABL1 kinase domain A-loop, P-loop, ATP-binding site, and substrate-binding pocket account for the majority of mutations detected in patients on TKI therapy, scores of other, distinct BCR-ABL1 mutations have also been detected [Soverini et al. 2011b].
Following second-line therapy
In patients treated with first-line imatinib followed by second-line nilotinib or dasatinib, evidence shows that patients who harbor BCR-ABL1 mutations conferring resistance to first-line imatinib are more likely than patients without such mutations to develop additional mutations on second-line TKI therapy [Shah et al. 2007; Soverini et al. 2009]. This implies that the number of mutations that might develop increases with the number of lines of TKI therapy, although this relationship has not been thoroughly examined. Furthermore, the array of mutations observed to occur in these patients is narrow and nonoverlapping, with the exception of T315I, which is detected frequently in patients with resistance to second-line nilotinib or dasatinib. Mutations primarily affecting Y253 and E255 (P-loop) are common following second-line nilotinib [Soverini et al. 2009], and mutations affecting V299 or F317 (ATP-binding region) are common following second-line dasatinib [Shah et al. 2007; Soverini et al. 2009]. These data strongly support the hypothesis described earlier that TKI treatment leads to selection of leukemic cell clones with specific BCR-ABL1 mutations.
As clinical experience accrues with second-line bosutinib and ponatinib after first-line imatinib, nilotinib, or dasatinib, information will emerge about the types of mutations selected for by second-line treatment with these newer TKIs.
Practical considerations for BCR-ABL1 kinase domain mutational analysis
When should mutational analysis be done?
We agree with NCCN Guidelines that mutational analysis should be conducted in patients who fail to achieve first-line TKI treatment milestones (≤10% BCR-ABL1 transcript levels or partial cytogenetic response at 3 and 6 months, or CCyR at 12 and 18 months), who lose response [hematologic or cytogenetic relapse, or a ≥1-log increase in BCR-ABL1 and loss of major molecular response MMR)], or who experience progression of CML to AP or BP [NCCN, 2014] (Figure 3). In regard to patients with rising BCR-ABL1 transcript levels and loss of MMR, we recommend mutational analysis be done while the patient remains on their current TKI treatment, because the presence of underlying mutant cells may be masked by the proliferation of nonmutated BCR-ABL1 cells when kinase inhibition is stopped. Testing performed beyond the current NCCN recommendations is unlikely to be clinically beneficial.
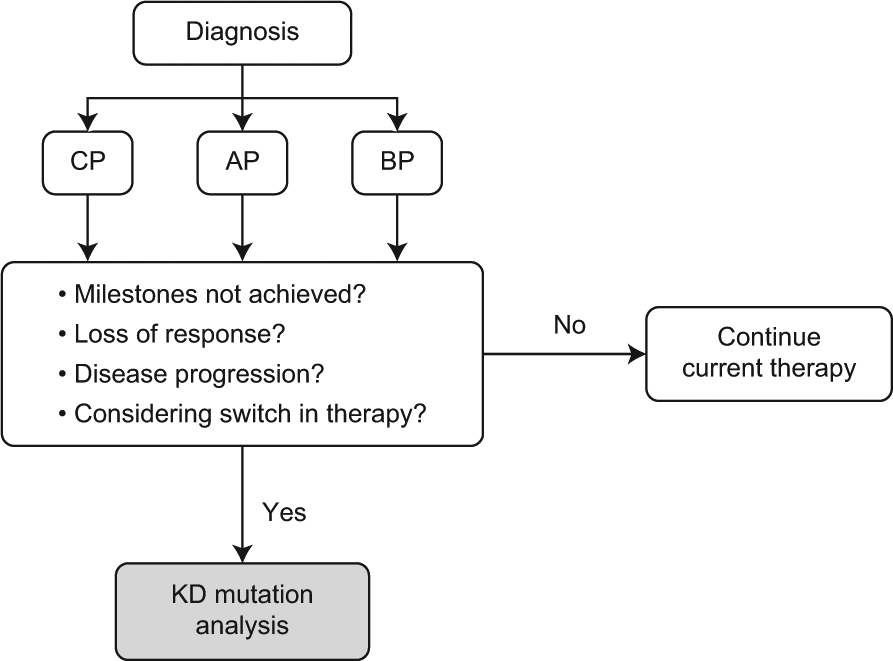
Algorithm for the use of BCR-ABL1 kinase domain mutational analysis in patients with chronic myeloid leukemia.
What type of testing should be done?
The NCCN Guidelines do not specify the method that should be used for mutational testing. The most common methods used for mutational analysis are summarized in Table 2. Direct sequencing remains the ‘gold standard’ for BCR-ABL1 mutational analysis because the technology is widely available and usually based on real-time polymerase chain reaction (PCR) amplification of BCR-ABL1 from RNA, a step that is already done for molecular monitoring [NCCN, 2014]. This method, however, is not sensitive enough to detect a mutant clone that accounts for less than 15–25% of total BCR-ABL1-positive cells [Hughes et al. 2006]. Other methods exist that are more sensitive than direct sequencing and can detect lower-level mutations. It is currently unclear what clinical significance, if any, low-level mutations may carry [Soverini et al. 2011a; Lange et al. 2013]. For example, in a study using ultra-deep sequencing (UDS) technologies to detect kinase domain mutations in patients treated with dasatinib, those with no mutations detectable by direct sequencing were subsequently found to have the F317I mutation at levels less than 15%. Despite their detection, the presence of this mutant clone at low levels did not affect response to treatment [Soverini et al. 2009], supporting the concept that response to TKI treatment is not perfectly predicted by mutation status alone. In other cases, mutations identified at baseline using highly sensitive techniques, namely allele-specific oligonucleotide (ASO)-PCR, have been detected by direct sequencing with denaturing high performance liquid chromatography (D-HPLC) a few months later [Ernst et al. 2009], suggesting that ASO-PCR might be more sensitive than direct sequencing with D-HPLC.
Methods for detection and quantification of BCR-ABL1 kinase domain mutations [Hughes et al. 2006; Alikian et al. 2012].

Not yet tested on chronic myeloid leukemia samples with mutant clones present at less than 1%.
How should results of mutational analysis be applied?
Like any other laboratory or test result, we consider the results of mutational analysis as one of many factors in making management decisions. Other factors we consider include drug efficacy and safety profile, patient comorbidities, and medical history. Because the action of a drug in a patient is infinitely more complex than interactions observed in cell culture, in vitro data alone are not generally reliable for predicting the clinical response of patients harboring a particular mutation to a specific TKI treatment. Supporting this concept, research indicates that factors other than in vitro sensitivity of mutations can predict clinical response (or resistance) to TKIs [Laneuville et al. 2010; Soverini et al. 2011b], such as drug pharmacokinetics and relative contribution of kinase domain mutations to the causes of clinical resistance [Soverini et al. 2011b].
Of the scores of BCR-ABL1 kinase domain mutations identified thus far in patients, there is a discrete set for which clinical experience has confirmed a relationship between in vitro and in vivo sensitivity to TKIs [Soverini et al. 2011b]. For this small number of mutations, the NCCN Guidelines suggest the following [NCCN, 2014]: dasatinib is recommended for mutations Y253H, E255K/V (P-loop), and F359V/C/I (substrate-binding region); nilotinib is recommended for mutations V299L, T315A, and F317L/V/I/C (ATP-binding region); bosutinib is recommended for mutations with Y253H, E255K/V, T315A, F317L/V/I/C, and F359V/C/I; ponatinib or omacetaxine are recommended for mutation T315I. (Please refer to current NCCN Guidelines for additional discussion of recommended therapies for these common mutations.)
Ponatinib is the only TKI that can bind to the T315I-mutant BCR-ABL1 kinase. In a phase I study of ponatinib in patients with refractory CML or Ph+ acute lymphoblastic leukemia, 12 of 12 patients with T315I mutation achieved CHR and 11 of 12 (92%) patients achieved major cytogenetic response (MCyR) on ponatinib [Cortes et al. 2012a]. Furthermore, 12-month follow up of a phase II study of ponatinib in patients with TKI resistance or intolerance or T315I showed that 45 of 64 (70%) patients with T315I achieved MCyR [Cortes et al. 2012b].
Omacetaxine is not a TKI but an inhibitor of protein synthesis (Synribo; Teva Pharmaceuticals USA, Inc., North Wales, PA, USA). Because its mechanism of action is not dependent on binding to BCR-ABL1, kinase mutants remain sensitive to omacetaxine. In a phase II study of omacetaxine in patients with T315I whose condition had failed to respond to prior TKI therapy, 48 of 62 (77%) patients achieved CHR, 14 (23%) achieved MCyR, and 10 (16%) achieved CCyR [Cortes et al. 2012c].
Is mutational analysis justifiable?
We can estimate the failure rate that is potentially attributable to the emergence of BCR-ABL1 mutations in patients with CML who initiate therapy with imatinib. A total of 5920 new cases of CML are estimated to be diagnosed in 2013 [American Cancer Society, 2013], and about 52% of newly diagnosed patients with CML receive first-line imatinib [Kantarjian et al. 2013]. Assuming 30–40% of patients on first-line imatinib develop resistance, and 15–50% of patients with imatinib resistance have BCR-ABL1 mutations, then newly emerging mutations are expected to occur in about 5–20% of patients who start on first-line imatinib. In about 57% of patients with mutations second-line TKI therapy fails, and in about 75% of patients with mutations, third-line TKI therapy fails [Soverini et al. 2009]. Thus, in about 2–9% of patients who start on first-line imatinib, TKI therapy may ultimately fail because of BCR-ABL1 mutations. The question then remains whether it is worthwhile to conduct mutational analysis in 30–40% (n = 924–1231) of patients with first-line imatinib resistance to identify the 2–9% (n = 59–263) of patients whose condition might ultimately fail to respond to TKI therapy.
Both the emergence of BCR-ABL1 kinase domain mutations [Soverini et al. 2005] and the failure to achieve expected treatment response milestones at 3 months and 12 months after the start of first-line TKI therapy [Druker et al. 2006; Brummendorf et al. 2012; Hanfstein et al. 2012; Hochhaus et al. 2012; Marin et al. 2012; Saglio et al. 2012] predict significantly worse long-term survival outcomes. Therefore, the detection of mutations in patients with resistance to first-line TKI therapy could identify those who might benefit from closer follow up or a change to more intensive therapy.
Again, whether or not to perform mutational analysis should be a highly individualized decision in which physicians should weigh multiple factors, such as type of resistance (incomplete response versus clear disease progression), sensitivity and cost of available mutation test methods, alternative treatment options, and more importantly, the likelihood of change in management if mutations are detected. For example, kinase domain mutation analysis may be more meaningful for a patient without a suitable stem cell donor who starts to lose response to a second-generation TKI and is in need of next-step decisions than it may be for a patient with a fully matched sibling donor who progresses to BP on imatinib.
Summary and conclusion
The development of resistance to a TKI calls for therapeutic intervention to avoid likely long-term negative effects on clinical outcome stemming from the lack of response, loss of response, or disease progression. Up to 20% of patients with CML who start on first-line TKI therapy will develop resistance to treatment that is caused by a mutation in the BCR-ABL1 kinase domain. Despite the importance of recognizing the signs of resistance and the use of BCR-ABL1 kinase domain mutational analysis to detection mutations, many clinicians do not fully appreciate the role of mutational analysis in the overall management of patients with CML. Current NCCN Guidelines recommend conducting BCR-ABL1 mutational analysis in patients with lack of response, loss of response, or disease progression. These are clearly the most appropriate clinical scenarios at this time in which to use kinase domain mutation testing for patients with CML. Most data showing the clinical importance of kinase domain mutations in CML were derived by direct sequencing; therefore the clinical relevance of low-level mutations detected by more sensitive techniques such as pyrosequencing, ASO–PCR, and UDS platforms is still unclear. The Interlaboratory Robustness of Next-generation Sequencing (IRON) II study is currently ongoing and aims to address this issue. Greater understanding of the practical utility of mutational analysis should shed light on this often underappreciated tool as a vital component of CML disease management.
Footnotes
Acknowledgements
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. We thank Anna Lau, PhD, and Claudette Knight, PharmD, of Percolation Communications LLC for their medical editorial assistance.
Funding
Novartis Pharmaceuticals Corporation provided financial support for manuscript development.
Conflict of interest statement
Ramon V. Tiu, MD, has been a member of the speaker’s bureaus for Novartis Corp., Bristol-Myers Squibb Co., and Teva Pharmaceutical Industries Ltd / Cephalon, Inc.
Jing Ai, MD, declares no conflicts of interest.