
Abstract
Atypical chronic myeloid leukemia (aCML) BCR-ABL1 negative is a rare myelodysplastic syndromes/myeloproliferative neoplasm (MDS/MPN) for which no standard treatment currently exists. The advent of next-generation sequencing has allowed our understanding of the molecular pathogenesis of aCML to be expanded and has made it possible for clinicians to more accurately differentiate aCML from similar MDS/MPN overlap syndrome and MPN counterparts, as MPN-associated driver mutations in JAK2, CALR, or MPL are typically absent in aCML. A 55-year old male with main complaints of weight loss and fatigue for more than half a year and night sweats for more than 2 months was admitted to our hospital. Further examination revealed increased white blood cells, splenomegaly, and grade 1 bone marrow fibrosis with JAK2 V617F, which supported a preliminary diagnosis of pre-primary marrow fibrosis. However, in addition to JAK2 V617F (51.00%), next-generation sequencing also detected SETBP1 D868N (46.00%), ASXL1 G645fs (36.09%), and SRSF2 P95_R102del (33.56%) mutations. According to the 2016 World Health Organization diagnostic criteria, the patient was ultimately diagnosed with rare aCML with concomitant JAK2 V617F and SETBP1 mutations. The patient received targeted therapy of ruxolitinib for 5 months and subsequently an additional four courses of combined hypomethylating therapy. The patient exhibited an optimal response, with decreased spleen volume by approximately 35% after therapy and improved symptom scores after therapy. In diagnosing primary bone marrow fibrosis, attention should be paid to the identification of MDS/MPN. In addition to basic cell morphology, mutational analysis using next-generation sequencing plays an increasingly important role in the differential diagnosis. aCML with concomitant JAK2 V617F and SETBP1 mutations has been rarely reported, and targeted therapy for mutated JAK2 may benefit patients, especially those not suitable recipients of hematopoietic stem cell transplants.
Background
The 2016 World Health Organization (WHO) category of myeloproliferative neoplasms (MPNs) includes three major subcategories of Janus kinase 2 (JAK2)/calreticulin (CALR)/thrombopoietin receptor gene (MPL) mutations related to MPNs, essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). 1 MPNs are usually distinguished from both myelodysplastic syndromes (MDS) and MDS/MPN due to the absence of morphologic dysplasia, which includes dysgranulopoiesis, dyserythropoiesis, and monocytosis. 2 Atypical chronic myeloid leukemia (aCML) is a rare kind of MDS/MPN. According to WHO, the current diagnostic criteria of aCML BCR-ABL1 negative indicate an evolution of classifying diseases that showed morphologic similarity to chronic myeloid leukemia (CML) but lacked both the Philadelphia chromosome (Ph chromosome) based on standard cytogenetics and BCR-ABL1 rearrangement based on polymerase chain reaction analysis. 1 Not only aCML, but also chronic neutrophilic leukemia (CNL), chronic myelomonocytic leukemia (CMML), and MDS/MPN are included in the differential diagnosis of these BCR-ABL1 negative hematologic neoplasms.1,3–6
Some cases of aCML have been named “CML-like syndrome” because both aCML and CML exhibit similar bone marrow with a spectrum of myeloid immaturity, including hyperplastic myeloid hyperplasia and peripheral blood leukocytosis. However, on morphologic grounds, that is where the similarity ends. Atypical CML is characterized by prominent dysplastic granulopoiesis, which is different from BCR-ABL1-positive CML. 1 Other features of aCML include the absence or minimal presence of monocytosis (10% of leukocytes) and basophilia (2% of leukocytes), which can help distinguish aCML from BCR-ABL1-positive CMML and CML, respectively.
The WHO-based laboratory criteria for MPNs has been complemented by knowledge gained from next-generation sequencing, and such knowledge also provides greater specificity in distinguishing aCML from MDS/MPN or MPNs.1,4,6,7 Because druggable targets may be unmasked, we should invariably focus on standard cytogenetic analysis along with myeloid mutation testing results at diagnosis and treatment of aCML. Over the past few years, characterization of the aCML mutational profile has expanded greatly. To our knowledge, MPN driver mutations include JAK2, CALR, and MPL variants, but JAK2 V617F has been described in only 4–8% of aCML.7,8 However, mutations of JAK2 along with mutations of SET binding protein 1 (SETBP1) and ethanolamine kinase 1 (ETNK1) are also known to be driver mutations in the pathogenesis of aCML and are useful in differentiating MPN from MDS/MPN overlap syndromes.9–11
Generally, aCML is highly associated with poor prognosis and a high chance of evolving into acute myeloid leukemia (AML). 10 While rare, aCML is an aggressive MDS/MPN with no current standard of care treatment. Allogeneic hematopoietic stem cell transplantation (HSCT) offers the only potential curative option. Therefore, allogeneic HSCT should always be a first consideration for eligible patients. Treatment strategies used for MDS and MPN, including targeted therapy and hypomethylating agents, have been largely relied on as nontransplant approaches for treating aCML. Although uncommon, the identification of JAK2 V617F or granulocyte-colony stimulating factor 3 receptor (CSF3R) T618I in aCML provides an opportunity to consider JAK inhibitor therapy, as both these mutations result in JAK-STAT pathway activation.
The patient in our current report refused our advice of allogeneic HSCT and opted for a lower-risk therapy. In view of a clear targetable mutation, targeted therapy was considered. The challenges in diagnosing and treating aCML relate to its high rate of transformation to AML, heterogeneous clinical and genetic features, and historically poor survival rates. Here, we report a rare case of aCML BCR-ABL1 negative with JAK2 V617F and concomitant SETBP1 mutations. After treatment with JAK inhibitor ruxolitinib for 5 months and four subsequent combined courses of demethylation therapy, the patient demonstrated a successful response.
Case report
Case presentation
A 55-year-old male was admitted to the Department of Hematology of The First Affiliated Hospital of Guangzhou University of Chinese Medicine because of atony of both lower extremities and malaise for more than 6 months. The patient reported increased fatigue and thirst during the last 2 months and described perspiring during sleep. He also had an associated weight loss of 5 kg during 6 months, and fever. Physical examination revealed splenomegaly.
Investigations
Laboratory examination of peripheral blood showed hyperleukocytosis with white blood cell (WBC) counts of 38.22 × 109/l and a platelet (PLT) counts of 390 × 109/l, and serum biochemical analysis showed lactate dehydrogenase (LDH) levels of 1122 U/l. Morphological examination of bone marrow (BM) aspirate showed a thrombocytosis and hypercellular marrow with high degree of dysgranulopoiesis, including 0.5% myeloblasts, 3.5% promyelocytes, 9% myelocytes, 3% metamyelocytes, 10% eosinophils, 4.5% lymphocyte, 2% nucleated red blood cells, and 9.5% monocytes (Figure 1). Manual differential count of peripheral blood also revealed a granulocytosis (Figure 2). BM biopsy analysis revealed hypercellular marrow, including neutrophilic leukocytosis to 84% with left shift and immature granulocytes, mean counts of 6–8 megakaryocytes/high power field combined with cell enlargement, and a lack of karyolobism. Immunohistochemical analysis identified myeloperoxidase (MPO)+, CD71+, CD61+, CD117−, and CD34+/− cells. Meshinprotein staining was consistent with grade 1 BM fibrosis (Figure 3). Molecular analyses of BCR/ABL fusion gene was negative and positive for JAK2 V617F. Cytogenetic analysis revealed a normal karyotype (46, XY). A preliminary diagnosis of prefibrotic primary myelofibrosis (pre-PMF) was considered with other myeloid neoplasms needing to be excluded. Next-generation sequencing was performed, and the results identified the JAK2 V617F variant (51% mutations load) along with three additional gene variants, additional sex combs like 1 (ASXL1) G645fs (36.09%), SETBP1 D868N (46.00%), and splicing factor, arginine/serine-rich 2 (SRSF2) P95_R102del (33.56%). Considering the SETBP1 expression, we retested peripheral blood cell morphology and performed a manual differential count that revealed 65% neutrophils, 10% metamyelocyte, 10% myelocytes, 2% promyelocytes, 8% monocytes, 2% basophilic granulocytes, 6% eosinophilic granulocytes, and 3% lymphocytes. Dysgranulopoiesis demonstrated abnormal nuclear segmentation, with 31% neutrophilic granulocyte band forms and 31% nuclear hyper-segmentation. Based on patient medical history, peripheral blood cell morphology, BM pathology, and genetic testing, the most appropriate diagnosis was atypical chronic myeloid leukemia with the coexistence of JAK2, SETBP1, ASXL1, and SRSF2 variants.

BM cell morphology with hematoxylin-eosin staining at high magnification (100X) of the patient at the time of diagnosis, which showed a hypercellular marrow and high granular level that indicates high degree of dysgranulopoiesis.
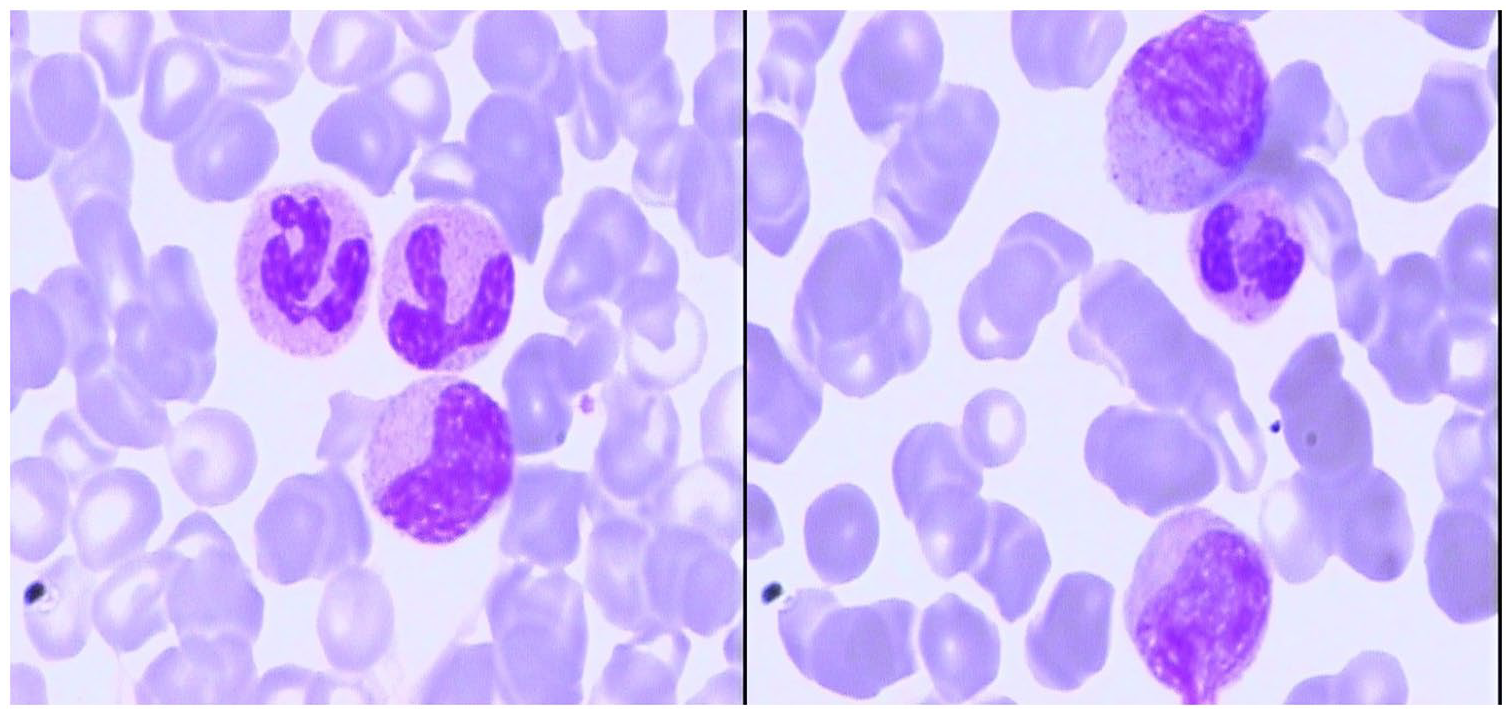
Blood smear at high magnification (100X) at the time of diagnosis: myelocytes and metamyelocytes could be found in peripheral blood with granulocytosis.

Immunohistochemical analysis of BM core biopsy at high magnification (200X) demonstrating CD71+ (panel 1) and CD61+ (panel 2). BM core biopsy with MPO+ (panel 3). Meshinprotein staining at high magnification (100X) demonstrating grade 1 BM fibrosis (panel 4).
Treatment
Considering the fact that allogeneic HSCT is the only potentially curative treatment for aCML, we recommended HSCT to the patient. However, taking into account the risks and costs of transplantation, the patient refused allogeneic HSCT and opted for treatment with lower risk and minimal pain. According to presence of the JAK2 mutation, a targeted therapy of ruxolitinib combined with symptomatic treatment was recommended since June 2019. For the first 5 months, the therapy consisted of ruxolitinib (40 mg/day). However, there was an aggravation of leukocytosis (from 38.22 g/l to 47.82 g/l), which, combined with the JAK2 mutation, motivated a change in treatment. Since 8 November 2019, Azacytidine (100 mg/day) was added to the treatment protocol daily for 7 days followed by 1× every 30 days. After four courses of therapy, the patient demonstrated a positive response, with effective symptomatic control and small improvement of thrombocythemia (from 368 × 109/l to 208 × 109/l). The most significant evolution was marked reduction in spleen size (thickness from 68 mm to 51 mm; length diameter from 173 mm to 145 mm) and decreased WBC counts from 38.22 × 109/l to 10.79 × 109/l. The JAK2 V617F mutations load dropped from 51% to 44.4%. However, the WBC counts rose again after the fourth course of azacytidine in March this year. A comprehensive follow-up examination on 18 March suggested that white blood cells are 15.54 × 109/l, hemoglobin is 139g/l, and PLT count is 248× 109/l. The results of BM and peripheral blood smears indicate that the granulocyte system is actively proliferating, accounting for 59.5% of nuclear cells. Peripheral blood neutrophil revealed a nucleus left shift, including 7% myelocyte, 6% metagranulocyte, 12% segmented neutrocyte, and 24% band form neutrophilic granulocyte. Eosinophils and basophils are easy to see. BM pathology results revealed that BM fibrosis has progressed to grade 3. Two additional gene mutations of NRAS and KRAS were found through another second-generation sequencing. And the JAK2 V617F mutations load raised to 46.2%. The spleen was more swollen than before, the long diameter increased to 168mm, and the thickness increased to 65mm. The above examination results indicate that the disease has progressed.
Discussion
Differential diagnosis
The central distinguishing feature in the 2016 WHO system for classification of tumors of the hematopoietic tissues is morphology. 11 Typically, aCML BCR-ABL1 negative is a rare form of MDS/MPN with poor overall patient survival.1,2,12 We report a case of aCML characterized by the absence of BCR-ABL transcripts and absolute monocytosis, with monocytes making up 10% of all leukocytes. In the current case, the patient was diagnosed with JAK2, ASXL1, SETBP1, and SRSF2 variants. The detection of MPN-related mutations, such as CSF3R, CALR, MPL or JAK2 mutations should prompt a differential diagnosis of PMF or MPN-U, which may share overlapping features with aCML. 13 It is more imperative to distinguish aCML from immature granulocytosis induced by other MPNs, especially pre-PMF. MDS/MPN cases are defined by the presence of both proliferative and dysplastic phenotypic features. 14 A key point to emphasize is a definite diagnosis in the presence or absence of ineffective hematopoiesis and granulocytic dysplasia (abnormal nuclei segmentation, abnormal chromatin clumping, or reduced or abnormal cytoplasmic granularity).15,16 The characteristic feature of pre-PMF is megakaryocytic proliferation and atypia without prominent immature granulocytosis and granulocytic dysplasia. 1 In addition, according to the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia, our diagnosis of aCML was supported by the presence of a SETBP1 variant as the MDS/MPN overlap category differs from MPN by a higher incidence of SETBP1 mutations (overall 25–30%).1,17 The current case is a reminder that a diagnosis should be based strictly on morphologic evaluation of blood and marrow as there are no defining mutations in aCML. Diagnostic criteria may be further refined in the future through more comprehensive molecular profiling to include genetic characteristics, which will result in decreased dependence on absolute cutoffs.
Molecular pathogenesis
Cytogenetic abnormalities are commonly associated in as many as 80% of cases aCML; however, there are currently no specific recurring cytogenetic findings that define aCML. 8 Driver mutations in aCML have been reported in CALR, CBL, CSF3R, ETNK1, JAK2, KIT, MPL, and RAS, but these drivers are neither specific to aCML nor present in the majority of aCML cases.18–20 Notably, co-expression of JAK2, ASXL1, SETBP1, and SRSF2 variants was described in only our case of aCML.
JAK2
A number of studies suggested that the JAK2 V617F mutation is the primary abnormality driving myeloproliferative neoplasms, 21 thus the JAK2 V617F mutation is frequent in PV, essential thrombocythemia (ET), and myelofibrosis (MF). 22 A guanine-to-thymine mutation at position 617 (V617F) in the JH2 domain of JAK2, which encodes a valine-to-phenylalanine substitution, can cause altered transcriptional activity and constitutive activation of signal transduction pathways. 23 Clinical signs of aCML, such as splenomegaly, overall survival, and transformation to AML, are related to JAK2 mutational status and allele burden.24,25 However, it remains to be established that the mechanism of proliferative advantage through a presence of JAK2 V617F mutation affects the pathogenesis of aCML as this JAK2 mutation is a uncommon in aCML (4–8%). 10 There have been only five reported cases of an association between aCML and the JAK2 V617F mutation. Table 1 is a summary of the clinical characteristics of JAK2 V617F-positive aCML patients described in the literature to date. Clinical studies indicate that the JAK inhibitor ruxolitinib is able to improve overall survival and its activity may be related to improved performance status and reduction of splenomegaly. 26 This supports JAK2 inhibition as a promising strategy for treating patients with aCML.
Summary of characteristics for patients with JAK2 V617F-positive aCML.
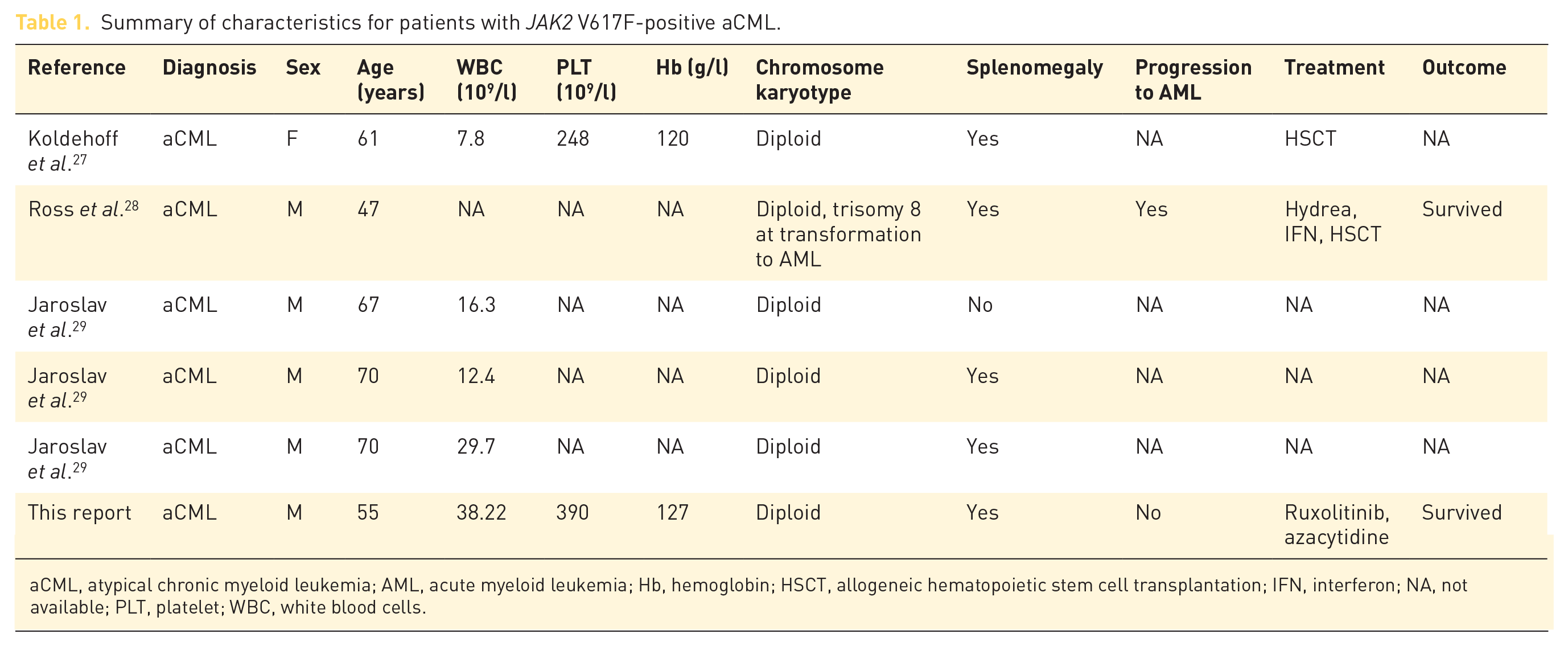
aCML, atypical chronic myeloid leukemia; AML, acute myeloid leukemia; Hb, hemoglobin; HSCT, allogeneic hematopoietic stem cell transplantation; IFN, interferon; NA, not available; PLT, platelet; WBC, white blood cells.
SETBP1
Mutated SETBP1 has shown predominance in aCML (10–48%). 30 However, the coexistence of mutations in SETBP1 and JAK2 in aCML has not been described to date. In fact, Meggendorfer et al. showed that SETBP1 and JAK2 V617F mutations were nearly mutually exclusive. 31 SETBP1, which is thought to be a negative regulator of tumor suppressor protein phosphatase 2A (PP2A), is expressed ubiquitously in adult human tissues. 32 Multiple research studies have also found that mutated SETBP1 in aCML patients is associated with a more adverse clinical profile related to higher leukocyte counts, lower hemoglobin levels, thrombocytopenia, and worse survival. 33 In some studies, SETBP1 variants with mutations such as the histone modification gene ASXL1 may have a cooperative function in leukemic progression of patients with aCML. 3 Therefore, SETBP1 analysis may constitute a valuable diagnostic tool in the prognosis of MDS/MPN syndromes, as individuals with SETBP1 mutations may have a worse prognosis than those with wild-type SETBP1.
SRSF2 and ASXL1
Mutational analyses showed that SRSF2 is frequently mutated in patients with aCML, with mutation frequencies of 12–40%. 34 SRSF2 and ASXL1 have been shown to be negative prognostic markers in MDS/MPN, which may also provide additional prognostic value in aCML.13,31 A multivariable analyses showed ASXL1 and SRSF2 mutations can independently be associated with poor prognostic rates. 35 A study reported by Manja et al. found a strong correlation between mutated SRSF2 and mutations in ASXL1 and SETBP1. 36 In this positive association, SETBP1 mutations are more often associated with SRSF2 mutations. In addition, SRSF2 mutations also often co-occur with mutated ASXL1. This indicates that ASXL1 and SETBP1 may have concomitant mutations with mutated SRSF2.
NRAS and KRAS
Approximately one-third of MDS/MPN cases were thought to present with mutations of NRAS and KRAS, which were initially considered as driver mutations of aCML.10,37 However, such mutations are also seen in a range of myeloid malignancies such as MDS, MPN, and even AML. Most of these mutations affect a range of essential, interrelated cellular mechanisms, including signalling, RNA splicing, transcriptional control, DNA damage response, and epigenetic regulation. 38 To our knowledge, inhibitors of these two signaling pathways may be effective as potential therapeutic targets. 39
The molecular findings described above are related closely to the pathogenesis of aCML, as well as the relationship between clinical phenotype and prognosis. The current case presented a diagnostic and therapeutic challenge, and demonstrated the importance of combining conventional karyotype/fluorescence in situ hybridization and next-generation sequencing technology to detect rare mutations in aCML.
Treatment of aCML
The challenges in managing aCML, a rare form of MDS/MPN, arise from the absence of robust randomized clinical trial data to support treatment recommendations. 40 The largest reported series of WHO-defined aCML cases consists of 55 cases from an Italian cohort who had an overall median survival of 25 months. 41 In this Italian cohort, transformation to AML occurred in 22 (40%) of patients, with the median time from diagnosis being 18 months. 41 Due to the poor prognosis of aCML, qualification for allogeneic HSCT ought to be the prioritized consideration in the treatment algorithm, because this is currently the only potentially curative treatment strategy for this disease according to three published representative retrospective analyses of transplantation series that focused on aCML.27,42,43 However, no recommendations on the optimal timing are provided.44,45 As with other cases of MDS/MPN, when HSCT is unavailable, the approach for treating MDS and MPN included the use of conventional chemotherapy and adjunctive agents aimed at disease cytoreduction. The use of hypomethylating agents in aCML may be a reasonable approach as their activity has been established from the treatment of MDS and CMML. Insights achieved from the mutational analysis of patients with aCML are currently promoting the investigation for several promising targeted therapies, including JAK inhibitor ruxolitinib, MEK inhibitor trametinib, and SRC kinase inhibitor dasatinib. Ruxolitinib is a JAK1/2 inhibitor currently approved by the United States Food and Drug Administration (FDA) for patients with intermediate-or high-risk myelofibrosis or those with polycythemia that is intolerant or resistant to hydroxyurea. Compared with the best available therapy, continuous ruxolitinib therapy is associated with durable and obviously reduction in splenomegaly and disease-related symptoms, improvement in quality of life, has only modest toxic effects, and may be consider as a bridge for patients with aCML that are eligible for allogeneic HSCT.46–48
Although our patient had a reduction in spleen volume of at least 35% and his WBC count was decreased significantly after treatment, we remain concerned regarding aCML-associated survival and his molecular profile. Specifically, there is an unfavorable prognosis associated with SETBP1, ASXL1, and SRSF2 variants in the context of other myeloid neoplasms. Based on the patient’s adverse clinical profiles, we speculated that he may ultimately have to confront a worse prognosis, and that there remains a high possibility of transformation to AML. If he unfortunately has to confront the worse prognosis, several protocols could be recommended to the patient. Firstly, we will still strongly recommend allogeneic HSCT as the only potential curative option. Moreover, we believe that therapy of subcutaneous injection of interferon is worth trying since the recombinant/pegylated interferon has proven efficacy for both newly diagnosed and previously treated patients with myeloproliferative neoplasms.49,50 On the other hand, recent second-generation sequencing revealed two gene mutations of NRAS and KRAS that may be associated with activation of the RAS-RAF-MAPK and PI3K signaling pathways, which may motivate a change of targeted therapy as some studies have indicated that the PI3K/Akt/mTOR pathway might represent a novel target for treatment in MPN. 51 If he finally unfortunately transforms to AML, the prognosis for a secondary AML will be very poor, as it often fails to respond to traditional chemotherapy. Advances in chemotherapy such as Vyxeos (CPX-351, liposomal daunorubicin: cytarabine) have proven overall benefit in secondary AML in fit candidates, and the addition of venetoclax to hypomethylating-based therapy in the unfit population seems to benefit both de novo and secondary AML. 52
Conclusion
We report an exceptional and rare case of aCML BCR-ABL1 negative. We have not identified any previous published reports regarding the coexistence of JAK2, ASXL1, SETBP1, and SRSF2 variants in patients with aCML. This report may be the first case in the literature describing this rare association in the same patient. Our findings increase the available information regarding the mechanisms by which malignancy arises, and will have important consequences for the diagnosis, prognosis, and treatment of patients with aCML and diseases associated with related gene mutations. However, the follow-up time for this patient is still short. The patient’s prognosis, including whether he will transform to leukemia, and how long it will take to transform, still needs longer follow up.
Footnotes
Acknowledgements
We would like to thank Editage for the language editing provided for this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the State Natural Sciences Fund, Project number 81903973; Guangdong Traditional Chinese Medicine Bureau, Project number 20202064; and Youth Fund Project for the Second Phase of the Innovation and Strong Hospital of the First Affiliated Hospital of Guangzhou University of Chinese Medicine, Project number 2019QN03.
Conflict of interest statement
The author(s) declare that there is no conflict of interest.
Ethics approval statement
This case report received exemption for ethical approval by The First Affiliated Hospital of Guangzhou University of Chinese Medicine. Ethics approval is not required for the publication of case reports because all patient-related data were collected retrospectively and anonymously.
Consent for publication
The provided information is de-identified and no health protected information is shared on this publication. Written informed consent to publish medical information and images was obtained from the patient reported in this publication.