
Abstract
Background:
GSK3326595 is a potent, selective, reversible protein arginine methyltransferase 5 (PRMT5) inhibitor under investigation for treatment of myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML), and acute myeloid leukemia (AML). In preclinical models of AML, PRMT5 inhibition decreased proliferation and increased cell death, supporting additional clinical research in myeloid neoplasms.
Objectives:
To determine the clinical activity, safety, tolerability, dosing, additional measures of clinical activity, pharmacokinetics, and pharmacodynamics of GSK3326595.
Design:
In part 1 of this open-label, multicenter, multipart, phase I/II study, adults with relapsed/refractory myeloid neoplasms (e.g., MDS, CMML, and AML) received monotherapy with 400 or 300 mg oral GSK3326595 once daily. Study termination occurred prior to part 2 enrollment.
Methods:
Clinical activity was determined by the clinical benefit rate (CBR; proportion of patients achieving complete remission (CR), complete marrow remission (mCR), partial remission, stable disease (SD) >8 weeks, or hematologic improvement). Adverse events (AEs) were assessed by incidence and severity. Exploratory examination of spliceosome mutations was performed to determine the relationship between genomic profiles and clinical response to GSK3326595.
Results:
Thirty patients with a median age of 73.5 years (range, 47–90) were enrolled; 13 (43%) and 17 (57%) received 400 and 300 mg of GSK3326595, respectively. Five (17%) patients met CBR criteria: 4 (13%) with SD >8 weeks and 1 (3%) achieving mCR. Of five patients with clinical benefit: three had SRSF2 mutation, one U2AF1, and one was splicing factor wild-type. Frequent GSK3326595-related AEs were decreased platelet count (27%), dysgeusia (23%), fatigue (20%), and nausea (20%). GSK3326595 had rapid absorption, with a Tmax of approximately 2 h and a terminal half-life of 4–6 h.
Conclusion:
GSK3326595 monotherapy had limited clinical activity in heavily pretreated patients despite robust target engagement. The safety profile was broadly consistent with other published PRMT5 inhibitor studies.
Trial registration:
ClinicalTrials.gov: NCT03614728.
Plain language summary
What is this study about? This summary provides the results of a study performed to see how safe and effective treatment with a once daily, oral medication called GSK3326595 was in patients with blood and bone marrow cancers.
What are PRMT5 inhibitors? GSK3326595 belongs to a class of medications known as PRMT5 inhibitors. PRMT5 is an enzyme that is involved in many processes in cells. In cancers, too much PRMT5 activity can cause excessive cell growth. This study was performed to see if blocking of PRMT5 by GSK3326595 would help treat patients with blood and bone marrow cancers.
What patients were in this study? The patients included in this study had previously received many other cancer treatments. Most patients with these types of cancers have few treatment options and usually pass away due to their disease.
What were the results? Five of the 30 patients (17%) included in the study had a response to treatment, including 4 patients with stable disease for more than 8 weeks and 1 patient with complete marrow remission for approximately 8 months. Of the 93% of patients that completed the study, 83% died. Ultimately, all 30 patients discontinued study treatment, mostly due to progression of their disease. The most frequent side effects related to GSK3326595 treatment that occurred in ⩾20% of patients were a decrease in the number of cells that help the blood clot, change in taste bud sense, fatigue, and nausea. The side effects caused by GSK3326595 were similar to what is seen with other PRMT5 inhibitors. Treatment with GSK3326595 provided limited benefits in this patient population and no future studies are planned for GSK3326595 at this time. Additional studies are needed for PRMT5 inhibitors, including combination therapies, to determine which patients with blood and bone marrow cancers could potentially benefit from treatment.
Keywords
Introduction
Myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML) are myeloid neoplasms with high risk of progression to acute myeloid leukemia (AML).1–5 Available chemotherapies for MDS and CMML yield meaningful responses in only a small minority of patients, and while allogeneic stem cell transplant is generally the only curative treatment, it is not always an option due to age, comorbidities, or donor availability.2,6–8 Most patients who respond to initial treatment will later progress, and, as effective salvage options are few, patients ultimately die of their disease. 2 The expected 5-year relative survival rates for MDS, CMML, and AML are 36.9%, 28.6%, and 31.9%, respectively.9–11
Identification of pathways involved in epigenetic and splicing dysregulation that affect proliferation and cell survival has led to novel therapeutic targets being explored as potential treatments for patients with myeloid neoplasms.2,7,8 One such potential target is protein arginine methyltransferase 5 (PRMT5), a member of the PRMT family of enzymes that methylate arginines in proteins, which are involved in many cellular processes, including precursor mRNA (pre-mRNA) processing, splicing, and transcriptional regulation.12–17 Overexpression of PRMT5 leads to induction of cellular hyperproliferation and is seen in several tumors, including myeloid malignancies.14–18 In preclinical models of AML, PRMT5 inhibition led to decreased proliferation and apoptosis, suggesting PRMT5 could be a therapeutic target for hematologic malignancies.18–20
PRMT5 accounts for most of the symmetric dimethylation of arginine in vivo,14,18 with plasma levels of symmetrical dimethylarginine (SDMA) serving as a surrogate pharmacodynamic (PD) biomarker of enzymatic PRMT5 activity, indicating the level of target engagement. 21 A correlation between SDMA reduction and tumor response has been shown in preclinical animal studies.20,21
Additionally, PRMT5 plays an important role in the splicing of mRNA via methylation of proteins in the spliceosome, the dysregulation of which can lead to an increase in aberrant splicing and downstream impact on target genes of spliceosome machinery.22–25 Somatic mutations in splicing factor genes are common in MDS and CMML, where approximately 60% of cases will harbor mutations in one of the core splicing factors: splicing factor 3B subunit 1 (SF3B1), serine/arginine-rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1), and zinc finger CCCH-type, RNA-binding motif and serine/arginine-rich 2 (ZRSR2).24,26 These appear to be mutually exclusive mutations (i.e., not more than one in each clone), suggesting that cells can only tolerate a certain degree of spliceosome dysfunction. In hematological malignancies, targeting of alternative splicing is an area of clinical interest. 27 Therapies that further alter pre-mRNA splicing (e.g., via pharmacologic inhibition of spliceosomal machinery) may provide therapeutic benefit to patients with myeloid neoplasms who harbor splicing factor mutations. 24
The primary objective of this first-in-class phase I/II trial was to assess the safety, pharmacokinetics (PK), PD, and preliminary clinical activity of GSK3326595, a potent, selective, reversible inhibitor of PRMT5, in patients with relapsed and/or refractory MDS, CMML, or AML.
Methods
Trial design
This was an open-label, multicenter, multipart, phase I/II study (ClinicalTrials.gov identifier: NCT03614728) to assess the safety, tolerability, PK, PD, and clinical activity of GSK3326595 in patients with relapsed and/or refractory MDS, CMML, or hypoproliferative AML. The study was conducted at seven centers (six in the United States and one in Canada) from October 2018 until April 2022. Additional trial design details are presented in the Supplemental Information.
Part 1 of the study consisted of a safety evaluation (dose confirmation), followed by a single-arm dose-expansion cohort to determine the clinical benefit rate (CBR) in patients treated with GSK3326595 monotherapy (Figure 1). Part 2 was planned to consist of two dose-escalation cohorts of GSK3326595 in combination with 5-azacitidine followed by a single-arm dose-expansion cohort. However, futility criteria were met during an interim analysis and the study was terminated prior to any patients being enrolled in part 2. Due to early termination, this report focuses on part 1 only.

Study design.
Eligibility criteria
Adults ⩾18 years of age with adequate organ function, an Eastern Cooperative Oncology Group performance status of ⩽2 and a diagnosis of either (1) intermediate, high, or very high-risk MDS as classified by International Prognostic Scoring System—Revised (IPSS-R) criteria; (2) intermediate or high-risk CMML per CMML-specific prognostic scoring system (CPSS) or clinical/molecular CPSS (CPSS-mol) criteria; (3) MDS or CMML secondary to prior antineoplastic therapy of any risk score; or (4) AML evolved from an antecedent MDS/myeloproliferative neoplasm of any risk score provided there was ⩽30% myeloblasts in the marrow or peripheral white blood cell (WBC) count was less than 20,000 cells/µL in the absence of leukoreduction therapy such as hydroxyurea or leukapheresis were eligible for the study. Patients were included if they had disease that failed to respond to, or progressed despite, treatment with at least one systemic therapy.
It was planned that at least 12 patients in part 1 were required to have documented loss-of-function mutation(s) in one or more of the following genes or proteins: SF3B1, SRSF2, U2AF1, or ZRSR2. In addition, documentation of the wild-type status of all of these genes/proteins was planned in a minimum of 12 patients to explore the potential utility of PRMT5 inhibition in patients with and without splicing factor mutations.
Full inclusion/exclusion criteria for part 1 are included in Supplemental Information.
Study treatment
Treatment schedule and starting dose were selected based on the recommended part 2 dose identified in a previous phase I study conducted in solid tumors and lymphoma (METEOR-1; NCT02783300). Originally, a 400-mg once-daily dosing was planned, and this was the starting dose for the dose-confirmation cohort. Due to a recommended dose change based on review of METEOR-1 study data, the dose-expansion cohort in this study was started at the lower dose of 300 mg once daily. The primary reason for this change was to optimize tolerability following review of data at the 400-mg dose in this and the METEOR-1 study. Dose escalation beyond 400 mg once daily was not planned unless emerging data indicated that a higher dose was appropriate (Figure 1).
GSK3326595 was orally administered once daily with water only, every day of each 28-day cycle, at approximately the same time of day ±4 h. On each PK collection day (days 1 and 15), samples were collected within 1 h prior to dosing.
Study endpoints
The primary endpoint was the CBR, defined as the percentage of patients achieving complete remission (CR), complete marrow remission (mCR), partial remission (PR), stable disease (SD) lasting for a period of at least 8 weeks, or hematologic improvement (HI), per International Working Group criteria. The incidence of dose-limiting toxicities (DLTs) was the primary endpoint for the 400-mg dose-confirmation cohort. A DLT was defined as an event that met predefined criteria, occurring within the first 28 days of treatment unless it was clearly established that the event was unrelated to treatment.
Secondary endpoints included overall response rate (ORR), progression-free survival (PFS), overall survival (OS), frequency and severity of adverse events (AEs), and PK and PD as exploratory endpoints. ORR was defined as the percentage of patients achieving CR, mCR, or PR per International Working Group criteria.
PD analyses
Symmetric dimethylarginine levels in blood and tumor samples were analyzed to explore the PDs of GSK3326595. Plasma samples were collected predose to measure SDMA using a mass spectrometry-based approach, and at 15, 29, 57, 91, and 169 days post-treatment.
Bone marrow biopsies were collected predose and 29 days after treatment. SDMA loss was measured in formalin-fixed paraffin-embedded tumor biopsies by immunohistochemistry (IHC). Additional PD analyses IHC details are available in Supplemental Information.
Spliceosome mutations were examined to determine if there was a correlation between baseline genomic profiles and clinical response to GSK3326595. Testing was performed locally, followed by central confirmation. Only centrally confirmed data was included in the formal analysis. Additional PD analyses splicing factor assessment details available in Supplemental Information.
Dose confirmation
Enrollment of a minimum of 6 and up to 12 patients was estimated for part 1 of the dose-confirmation cohort. If a patient was unable to receive at least 75% of intended doses within the 28-day DLT observation period for any reason other than a predefined DLT (e.g., concurrent illness or disease progression) in the 400-mg dose-confirmation cohort, the patient was replaced by an additional patient assigned to the same dose. Although no formal statistical hypotheses were tested, the part 1 dose confirmation was guided by the Neuenschwander continual reassessment method for dose-escalation/deescalation decisions if DLTs occurred.
Interim analysis
For the 300-mg dose-expansion cohort only, a first interim futility analysis was planned after a minimum of eight evaluable patients completed at least two postbaseline efficacy assessments, had disease progression or died, or permanently discontinued the study intervention. Subsequent interim futility analyses would be performed every 2–3 months, depending on enrollment, with the addition of a minimum of five evaluable patients. If the predictive probability of success was determined to be less than 1% (which equated to clinical benefit occurring in 0 out of 8 and ⩽3 out of 17 patients in the first and second interim analyses, respectively), the study would be terminated due to meeting futility criteria. Success was defined as the posterior probability of CBR >30% at the end of the cohort being larger than 89.1%. At the maximum sample size of 35 patients, the design had type 1 error of 0.07 and power of 83%, if the true CBR was 30% and 50%, respectively, using minimally informative prior of Beta (0.05, 0.05).
Statistical analysis
Statistical analyses were performed with SAS software version 9.4 (SAS Institute Inc., Cary, NC, USA). Safety and efficacy analyses were based on the All-Treated population, defined as all patients who received at least one dose of GSK3326595. The PK population consisted of all patients from the All-Treated population for whom a PK sample was gathered and analyzed.
The primary efficacy analysis was the percentage of patients meeting CBR criteria. The number of patients with best overall responses was summarized in the following response categories: CR, mCR, PR, SD ⩾8 weeks, SD <8 weeks, HI, CBR (CR + mCR + PR + SD ⩾8 weeks + HI), PD, and not evaluable. The corresponding exact two-sided 95% confidence interval (CI; Clopper-Pearson confidence limits for the binomial proportion) for CBR was also calculated.
Patients who received at least 75% of the planned doses of GSK3326595 within the 28-day DLT observation period or those who had a DLT were included in the DLT evaluable population. Additional statistical analysis details are available in Supplemental Information.
Data availability statement
Anonymized individual participant data and study documents can be requested for further research from https://www.gsk-studyregister.com/en/.
Results
Study disposition
Thirty patients were enrolled in study part 1, with 13/30 (43%) patients in the dose-confirmation cohort receiving 400 mg GSK3326595 and 17/30 (57%) patients assigned to the dose-expansion cohort receiving 300 mg GSK3326595. Twenty-eight (93%) patients completed the study (defined as death or study completion), of whom 25 (83%) died and 3 (10%) completed follow-up (Supplemental Table 1). Two patients withdrew from the study because of either termination of the study by sponsor (1 (3%)) or physician’s decision (1 (3%)). All 30 (100%) patients discontinued study treatment, mostly due to disease progression (13 (43%) patients). Additional discontinuation reasons were physician decision (6 (20%)), AE (4 (13%)), other (6 (20%)), and patient withdrawal (1 (3%)).
Patient population and treatment exposure
Patients had a median age of 73.5 years (range, 47–90), with the majority being male (23 (77%); Table 1), and an equal number of patients with an initial diagnosis of AML or MDS (14 (47%) patients each) and 2 (7%) patients with CMML. All patients with MDS or CMML had a risk category of intermediate or higher based on the IPSS-R prognostic risk score and CPSS molecular risk scoring system, respectively. Patients with AML were categorized based on World Health Organization classification criteria. Twelve (40%) patients had one or more gene mutations identified at screening, including mutations in spliceosome complex genes (SRSF2, SF3B1, U2AF1), tumor suppressors (TP53), regulators of chromatin remodeling and gene expression (additional sex combs-like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), runt-related transcription factor-1 (RUNX1)), tyrosine kinases fms-like tyrosine kinase 3- internal tandem duplication (FLT3-ITD), and other mutations (B-cell lymphoma 6 corepressor (BCOR), colony stimulating factor 3 receptor (CSF3R), SET binding protein 1 (SETBP1)). Nine patients (30%) had no identified mutation at screening, and nine patients (30%) had unknown mutation status. All 30 (100%) patients had prior anticancer therapy. The most common prior treatment was chemotherapy (30 (100%)), and 4 (13%) patients had received prior allogeneic stem cell transplant. This population received a wide range of prior chemotherapy treatments including hypomethylating agents alone (e.g., azacitidine, decitabine) or in combination with venetoclax, 7 + 3 regimen (cytarabine and daunorubicin), MEC (mitoxantrone, etoposide, and cytarabine), CLAG-M (cladribine, cytarabine, G-CSF, and mitoxantrone) in combination with midostaurin, and FLAG-Ida (e.g., fludarabine, cytarabine, G-CSF, and idarubicin).
Baseline patient demographics and disease characteristics.

AML, acute myeloid leukemia; BMI, body mass index; CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndrome.
The thirty patients received GSK3326595 for a median of 1.15 months (range, 0.5–12.9), with a median daily dose of 300 mg (range, 157–400 mg; Supplemental Table 2).
Clinical activity
Overall, 5/30 (17%) patients met CBR (95% CI: 5.6, 34.7): 2/13 (15%) in the 400-mg cohort (95% CI: 1.9, 45.4) and 3/17 (18%) in the 300-mg cohort (95% CI: 3.8, 43.4; Table 2). Of these, 4/30 (13%) patients had SD for more than 8 weeks (two patients each in the 400- and 300-mg cohorts), and 1/30 (3%) patients, who were in the 300-mg cohort, had mCR. Two of the five patients with a protocol-defined clinical benefit of SD >8 weeks were treated in the 400-mg cohort (one patient with a diagnosis of AML and one patient with MDS) until weeks 24 and 25 of the study, respectively. Of the three patients who experienced CBR in the 300-mg cohort, one patient with MDS and one with AML had the best response of SD >8 weeks and were treated for 14 and 12 weeks, respectively. One patient with MDS had mCR for 245 days and was treated for >40 weeks. Additional characteristics of patients meeting CBR criteria are shown in Supplemental Table 3.
Summary of clinical response per International Working Group criteria.

CBR, clinical benefit rate; CI, confidence interval; CR, complete response; HI, hematologic improvement; mCR, complete marrow remission; ORR, overall response rate; PR, partial remission; SD, stable disease.
CBR defined as CR + mCR + PR + SD ⩾8 weeks + HI.
CI are based on Clopper-Pearson confidence limits.
ORR defined as CR + mCR + PR.
Thirteen (43%) patients had disease progression while on study treatment. Seven (23%) patients were not evaluable: four patients from the 400-mg cohort and three patients from the 300-mg cohort. Patients were categorized as not evaluable if they lacked a postbaseline study assessment at the time of study withdrawal.
In the 400-mg cohort, there were no responders (95% CI: 0.0, 24.7), while 1 (6%) patient in the 300-mg cohort achieved ORR by mCR (95% CI: 0.1, 28.7; Table 2).
Median PFS was similar in both cohorts; 0.99 months (95% CI: 0.95, 2.53) in the 400-mg cohort and 0.99 months (95% CI: 0.95, 2.43) in the 300-mg cohort. Fourteen (47%) of the 30 patients experienced disease progression and 11 of the 30 patients (37%) died during the study. Patients in the 400- and 300-mg cohorts had a median OS of 2.69 (95% CI: 0.99, 12.98) and 2.96 (95% CI: 1.18, 15.24) months, respectively. Overall, there were no clinical differences in efficacy between patients in the two cohorts.
Interim analysis outcome
Two interim analyses were performed in the 300-mg dose cohort only, when 8 and 17 patients were evaluable. Following the second interim analysis review of the 300-mg part 1 cohort, from a June 30th, 2020 data cutoff, futility criteria were met and the 300-mg cohort was stopped.
PK and PD
Absorption of GSK3326595 was rapid, with a Tmax of approximately 2 h and a terminal half-life of 4–6 h (Supplemental Table 4). Minimal accumulation of GSK3326595 following repeated daily administration occurred, and GSK3326595 exposure was dose-proportional. Mean and median concentration-time plots for the 300- and 400-mg cohorts on days 1 and 15 are shown in Figure 2 and Supplemental Figure 1.

GSK3326595 mean concentration-time plots by treatment and visit.
After 15 days of treatment, there was a mean (standard deviation) percentage decrease from baseline in plasma SDMA of 75.6% (15.1%) in the 400-mg dose cohort (N = 10) and 77.7% (5.0%) at 300 mg (N = 17; Figure 3). Maximum PD effect was reached by day 29, with an 84.1% (4.3%) and 83.7% (3.1%) reduction in the 400- and 300-mg cohorts, respectively. No additional reduction occurred at later time points.
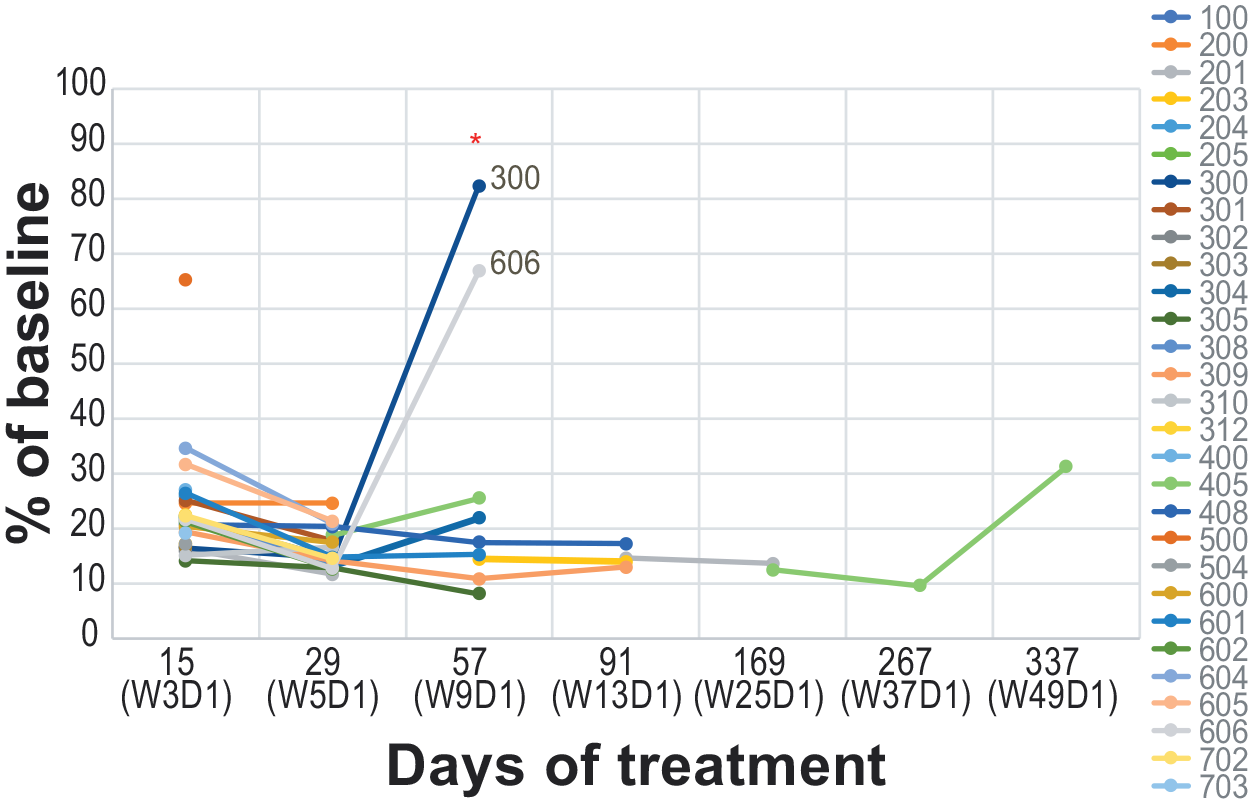
Plasma SDMA in patients treated with GSK3326595.
The level of reduction in plasma SDMA data correlated with tumor SDMA data in 11 evaluable samples: 4 patients in the 400-mg cohort and 7 patients in the 300-mg cohort. After 29 days of treatment, there was a mean (standard deviation) percentage decrease in SDMA observed in bone marrow biopsies of 85.75% (3.14%) in the 400-mg cohort and 84.63% (2.9%) in the 300-mg cohort. The maximum observed decrease in tumor SDMA was 100% and 97.8% at the same time point for the 400- and 300-mg cohorts, respectively.
DNA was isolated and sequenced from both blood and bone marrow aspirates for each patient to determine the presence of splicing factor mutations. Due to one patient having a bone marrow sample with volume too low to obtain sequencing from, blood samples were utilized instead of bone marrow to include this patient in the results. Concordance was almost 100% when blood and bone marrow data were compared. Ten of the 19 evaluable patients harbored centrally confirmed spliceosome mutations (5 had SF3B1, 4 SRSF2, and 1 U2AF1). Of those 10 patients, 2 (20%) patients with centrally confirmed hotspot mutations in the SRSF2 gene showed clinical benefit (both patients had MDS and the best response of SD, treated until weeks 25 and 14, respectively) from treatment with either a 400- or 300-mg daily dose of GSK3326595, respectively. Two additional patients were identified locally as having a spliceosome mutation: U2AF1 in one patient with AML and SRSF2 in one patient with MDS. Both patients had clinical benefits, but these were not included in the formal analysis as they were not confirmed centrally (sample was not collected due to lack of patient consent); the one patient with AML in the 400-mg cohort had SD and was treated until week 24, and the one patient with MDS in the 300-mg cohort had mCR and was treated for >40 weeks. Of the nine patients who were assessed centrally as having no evidence of splicing factor gene mutations, 1 (11%) patient with AML met the criteria for SD and was treated until week 12 with a 300-mg daily dose of GSK3326595. With respect to local testing of splicing factor mutations, 8 patients had spliceosome mutations, 13 patients did not have a loss-of-function mutation, and the mutational status of the remaining 9 patients was unknown. Among these locally assessed patients, 4/8 (50%) with spliceosome mutations (3 SRSF2, 1 U2AF1) and 1 patient (8%) without a splicing factor gene mutation had clinical benefit from treatment with GSK3326595. Concordance analysis between central and local testing of splicing factor mutations was conducted and included 16 of the 19 patients that were tested centrally. By local assessment, 3 of the 19 patients had an unknown splicing mutation status, resulting in their exclusion from the concordance analysis. The concordance of central versus local testing was 81.25% (13/16 concordant). All splicing factor mutations detected by local testing that had central testing performed were concordant.
Safety
The DLT evaluable population consisted of 26 patients (10/13 (77%) patients in the 400-mg cohort and 16/17 (94%) patients in the 300-mg cohort). No DLTs occurred in either cohort during the 28-day observation period.
All patients in part 1 of the study (N = 30) experienced a treatment-emergent adverse event (TEAE; Table 3). The most frequent TEAEs were diarrhea and fatigue (13 (43%) patients each), decreased platelet count (12 (40%)), dyspnea (11 (37%)), anemia and dysgeusia (10 (33%) patients each), and decreased appetite and nausea (9 (30%) patient each). The remaining TEAEs were reported in less than 30% of patients, including thrombocytopenia, which occurred in 5 (17%) patients.
Treatment-emergent adverse events.
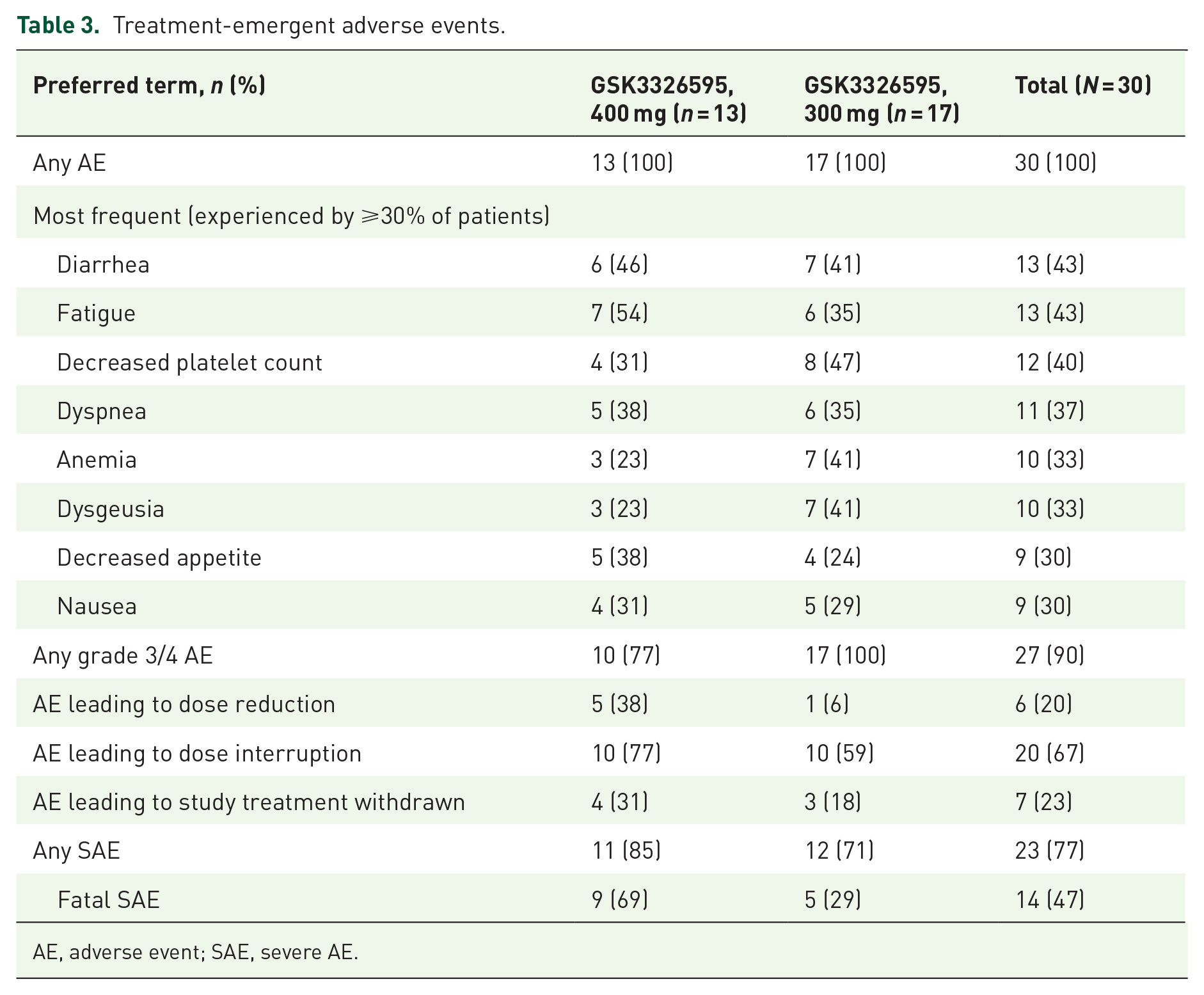
AE, adverse event; SAE, severe AE.
The most frequent GSK3326595-related TEAEs (occurring in ⩾10% of patients) were decreased platelet count (8 (27%)), dysgeusia (7 (23%)), fatigue and nausea (6 (20%) patients each), and decreased appetite and neutrophil count (4 (13%) patients each; Table 4). Thrombocytopenia and anemia were reported in 3 (10%) patients each and were attributed to GSK3326595 treatment.
GSK3326595-related treatment-emergent adverse events.

AE, adverse event; SAE, severe AE; TEAE, treatment-emergent AE.
Grade 3/4 TEAEs occurred in 10 (77%) and 17 (100%) patients in the 400- and 300-mg cohorts, respectively. Decreased platelet count (11 (37%)), anemia (8 (27%)), decreased neutrophil count (7 (23%)), pneumonia and febrile neutropenia (6 (20%) patients each), thrombocytopenia (5 (17%)), fatigue, hypotension, and dyspnea (4 (13%) patients each) were the most frequent TEAEs reported with a maximum grade of 3 or 4. GSK3326595-related grade 3/4 TEAEs were reported in 5 (38%) patients in the 400-mg cohort and 8 (47%) patients in the 300-mg cohort. Decreased platelet count (6 (20%)), decreased neutrophil count (4 (13%)), and thrombocytopenia (3 (10%)) were the most frequent TEAEs reported with a maximum grade of 3 or 4.
Serious adverse events (SAEs) occurred in 23/30 (77%) patients (11/13 (85%) and 12/17 (71%) receiving 400- and 300-mg of GSK3326595, respectively). The most common SAEs (occurring in ⩾10% of patients) were sepsis (8 (27%)), febrile neutropenia (5 (17%)), pneumonia (4 (13%)), anemia and thrombocytopenia (3 (10%) patients each). Treatment-related SAEs occurred in 6 (20%) patients, with decreased neutrophil count being the most reported in 3 (10%) patients. No other treatment-related SAE was reported in more than one patient each.
Study treatment was discontinued due to TEAEs in 7/30 (23%) patients, with sepsis being the most frequently reported cause of discontinuation (n = 2, 7%). All other TEAEs leading to discontinuation occurred in one patient each and included interstitial lung disease, cellulitis, multiple organ dysfunction syndrome, septic shock, subdural hematoma, and increased WBC count.
A total of 25 patients (83%) died during the study, 11 (85%) in the 400-mg cohort and 14 (82%) in the 300-mg cohort. Fourteen (47%) deaths occurred ⩽30 days from last dose of study treatment (6 in the 400-mg and 8 in the 300-mg cohort) and 11 (37%) deaths occurred >30 days from last dose of study treatment (5 in the 400-mg and 6 in the 300-mg cohort). A total of 9 (69%) patients in the 400-mg cohort and 5 (29%) patients in the 300-mg cohort experienced a fatal SAE. The most frequently reported fatal SAE in both treatment cohorts was sepsis. One participant with AML in the 300-mg cohort had a fatal SAE of sepsis, possibly related to study treatment. At the time of death, the patient also had ongoing SAEs of decreased neutrophil count and diverticulitis that were considered possibly related to GSK3326595 treatment.
Discussion
Preclinical data support PRMT5 as a potential therapeutic target in myeloid malignancies, and, to the best of our knowledge, this is the first clinical trial to report efficacy and safety data of a PRMT5 inhibitor in this setting. 18 The rationale for targeting PRMT5 in the treatment of MDS and CMML specifically is further supported because mutations in splicing factors occur in most patients with MDS and CMML, which may confer therapeutic vulnerability to PRMT5 inhibition.24,28,29 Despite promising preclinical data and evidence of robust target engagement demonstrated by SDMA suppression in the bone marrow and peripheral blood in this study, modest clinical activity was observed for GSK3326595 monotherapy. However, it is important to recognize that these patients had relapsed and/or refractory disease with a very poor prognosis, and only a single agent was employed. Combination therapy with a PRMT5 inhibitor and an HMA and/or venetoclax in spliceosome-mutant-only patients with myeloid malignancies has not yet been explored.
Seventeen percent of patients met the primary efficacy endpoint of CBR. No patients achieved a clinical response of CR or PR. One patient had mCR and four patients had SD for more than 8 weeks.
Only 2/10 patients with centrally confirmed splicing factor gene mutations had evidence of clinical benefit, arguably displaying limited evidence of a correlation with this potentially predictive biomarker in the context of this study. However, both of these patients had MDS with a centrally confirmed SRSF2 mutation. On local analysis, which was concordant with central analysis, 4/8 (50%) patients with a splicing factor mutation (3 SRSF2, 1 U2AF1) and 1/13 (8%) patients without a splicing factor mutation had evidence of clinical benefit, suggesting a possible signal in SRSF2.
Study limitations included a small patient sample size and early study termination. Arguably, the heterogeneity of the study population, which included patients with diagnoses of relapsed/refractory AML, MDS, and CMML, may have impacted the evaluation of efficacy in patients with different disease subtypes and/or molecular profiles (i.e., splicing mutant-only), however, in the context of this first-in-class phase I/II study (phase II not conducted) we believe this remained appropriate. Despite some evidence of clinical activity, study part 1 met prespecified efficacy futility criteria at the second planned interim analysis, resulting in study termination prior to any enrollment in part 2. Whether the planned combination of GSK3326595 and 5-azacitidine in part 2 would have led to increased efficacy and favorable benefit-risk remains unclear, as does the potential for clinical benefit in selected populations (i.e., those with spliceosome mutations alone or within select specific mutation subsets). However, preclinical data supported the rationale for this approach, and combination with hypomethylating agents (or other agents) could be explored for other PRMT5 inhibitors in development.
The safety profile of GSK3326595 monotherapy was broadly consistent with what was anticipated in this patient population and with published data for PRMT5 inhibitors to date,30–33 with similar commonly occurring AEs (fatigue, anemia, and nausea) and grade ⩾3 treatment-related AEs (thrombocytopenia and neutropenia). There were no DLTs reported in either cohort. Study termination did not occur due to any urgent safety issues or new safety concerns.
A number of PRMT5 inhibitors have now entered clinical development, predominantly in the solid tumor setting, and have generally demonstrated modest monotherapy activity in early phase trials involving patients with advanced/metastatic or recurrent disease. 34 To the best of our knowledge, currently, only one other PRMT5 inhibitor (PRT543) is being investigated in a myeloid malignancy setting and no clinical data for this drug has been formally presented to date (ClinicalTrials.gov). PRMT5 inhibitors have shown limited activity in advanced malignancies as monotherapy and may require the development of combination treatments that target unique therapeutic vulnerabilities and allow for more time for epigenetic therapies to take effect.
More broadly, therapeutic targeting of alternative splicing remains an area of clinical interest in hematological malignancies. 27 Targeted degradation of the RNA splicing factor RBM39 (which is required for cell survival of malignant cells with splicing factor mutations) by E7820 has shown evidence of target engagement, but limited efficacy. 35 This target may potentially provide utility in combination with other novel therapies. There are ongoing splicing modulator studies of PRT1419, a myeloid cell leukemia-1 inhibitor, as monotherapy and in combination with azacitidine or venetoclax (an FDA-approved B-cell lymphoma 2 (BCL-2) inhibitor). 36 H3B-8800 is a splicing modulator of the SF3b complex that had previously displayed limited activity as a single agent in advanced myeloid disease, but is currently being investigated as monotherapy for the treatment of patients with lower-risk MDS with an SF3B1 mutation and has shown antileukemic efficacy in preclinical data.37,38 Synthetic lethality induction may require novel combination therapies, and it remains uncertain whether targeting PRMT5 for the treatment of myeloid neoplasms will ultimately demonstrate clinical utility in specific subsets of patients with splicing factor mutations. Lastly, it is possible that only specific splicing factor mutations (e.g., SRSF2), not splicing mutations as a class, will confer sensitivity to PRMT5 inhibition.
Conclusion
Despite encouraging preclinical data, and evidence of robust target engagement, clinical activity of GSK3326595 monotherapy at the 300- and 400-mg doses was limited in this patient population, which was heavily pretreated with poor prognosis. GSK3326595 exhibited a safety profile broadly consistent with published PRMT5 inhibitor data and anticipated in the study population. No future clinical trials for GSK3326595 are planned at this time; however, it will be important for drugs targeting PRMT5, or associated pathways, to focus on the identification of predictive biomarkers to monotherapy or combination treatment of patient subgroups most likely to benefit from PRMT5 inhibition, such as SRSF2-mutant myeloid malignancies.
Supplemental Material
sj-docx-1-tah-10.1177_20406207241275376 – Supplemental material for Phase I/II study of the clinical activity and safety of GSK3326595 in patients with myeloid neoplasms
Supplemental material, sj-docx-1-tah-10.1177_20406207241275376 for Phase I/II study of the clinical activity and safety of GSK3326595 in patients with myeloid neoplasms by Justin Watts, Mark D. Minden, Kimo Bachiashvili, Andrew M. Brunner, Sameem Abedin, Timothy Crossman, Magdalena Zajac, Veronica Moroz, Jacqueline L. Egger, Aarti Tarkar, Brandon E. Kremer, Olena Barbash and Gautam Borthakur in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Medical editorial support, Lacy Miron, PharmD (assembling tables and figures, collating author comments, copyediting, fact-checking, and referencing) and graphic services were provided by AOIC, LLC and were funded by GSK. Ipsen reviewed this manuscript for scientific accuracy but had no input into the content.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.