
Abstract
While diffuse large B-cell lymphoma (DLBCL) was once considered to be a single disease entity, recent biological insights have demonstrated that it can be divided up into at least three molecular subtypes. Gene expression profiling has revealed that DLBCL consists of a germinal center B-cell like subtype (GCB), an activated B-cell like subtype (ABC) and a primary mediastinal B-cell lymphoma subtype (PMBL). These three entities arise from different stages of B-cell differentiation and are characterized by distinct mechanisms of oncogenic activation. In GCB DLBCL, the BCL6 transcription factor may play an important role in tumor survival and treatment resistance and strategies that target this are under investigation. ABC DLBCL is characterized by high expression of target genes of the nuclear factor kappa B (NF-κB)/Rel family of transcription factors and strategies that target NF-κB are in clinical trials. PMBL is a distinct clinicopathologic entity that shares many molecular features with nodular sclerosis Hodgkin lymphoma (HL) and may benefit from dose intensity approaches and inhibition of the Janus kinases. Other biologic predictive factors such as MYC and BCL2 may be overexpressed in both the GCB and ABC subtypes and strategies that target these complexes are also being tested.
Keywords
Evolution in the classification and definition of diffuse large B-cell lymphoma
While earlier lymphoma classifications, like the National Cancer Institute Working Formulation, which was in use in the United States until the early 1990s lacked a biological foundation, later classifications such as the Kiel classification, which was used in Europe, and the Revised European-American Lymphoid (REAL) classification, published in 1994, were based on clinical-biological foundations [National Cancer Institute, 1982; Harris et al. 1994; Stansfeld et al. 1988]. More recently, in the 2008 World Health Organization (WHO) classification, major genetic and biological insights have been incorporated into the diagnostic criteria for lymphomas and for diffuse large B-cell lymphoma (DLBCL) [Jaffe, 2009]. This evolution in classification is the result of insights into the molecular pathogenesis and biology of lymphoma, including the identification of ‘hallmark’ genetic abnormalities and has paved the way for the discovery of driver pathways and clinical testing of targeted therapy [Tay et al. 2010; Wilson et al. 2010a].
While it has long been recognized that DLBCL is both clinically and biologically diverse, it had been challenging in the past to readily subdivide it into distinct disease entities because of overlapping morphologic and pathogenetic features [Jaffe et al. 2001]. As a result, treatment strategies have primarily depended on clinical features such as stage, age and the International Prognostic Index (IPI) score [The International Non-Hodgkin’s Lymphoma Prognostic Factors Project, 1993]. Large-scale gene expression profiling (GEP) and mutational analysis, however, has led to the recognition that DLBCL is a heterogeneous entity and can be divided into subtypes that arise from B cells at different stages of differentiation with distinctive molecular and clinical characteristics (Figure 1) [Jaffe, 2009]. Thus, when considering treatment and, in particular, when discussing novel therapies, it is essential to understand these pathobiological distinctions.

Oncogenic pathways for three subtypes of diffuse large B-cell lymphoma. On the basis of gene-expression profiling, diffuse large B-cell lymphoma can be divided into three molecular subtypes: the germinal-center B-cell-like (GCB) subtype, the activated B-cell-like (ABC) subtype, and primary mediastinal B-cell lymphoma (PMBL). These subtypes originate from various stages of B-cell differentiation and acquire distinct oncogenic abnormalities. The abnormalities that are listed are preferentially or exclusively observed in the indicated subtypes. Blue lines indicate activation and red lines indicate inhibition. AID, activation-induced cytidine deaminase; ITAM, immunoreceptor tyrosine-based activation motifs; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor κB. (Courtesy of Dr Louis Staudt, NCI.)
Molecular pathology of GCB and ABC DLBCL
In the most recent WHO classification, DLBCL is divided into four major groupings that are further divided along molecular, pathological and/or clinical grounds. Of these, the most common group is DLBCL not otherwise specified (NOS) that is further subdivided into the germinal center (GC) B-cell like subtype (GCB) and activated B-cell like subtype (ABC) molecular subgroups by GEP (Figure 2A) [Lenz et al. 2008b; Rosenwald et al. 2002]. In the initial GEP studies of DLBCL, arrays were performed on follicular lymphoma, chronic lymphocytic leukemia (CLL), lymphoma and leukemia cell lines, and normal lymphocyte subpopulations obtained under a variety of activation conditions to provide a comparative basis for analysis of DLBCL gene expression [Alizadeh et al. 2000]. Genes associated with cellular proliferation showed a clear distinction among the lymphoma types with DLBCL generally showing higher albeit variable expression [Wilson et al. 1997]. The proliferation signature genes were a diverse group and included cell-cycle control and checkpoint and myc genes. Another prominent feature of DLBCL was a group of genes that defined a ‘lymph-node’ signature that appeared to reflect the nonmalignant cells in the biopsy samples. Genes that distinguished GC B cells from other stages of B-cell differentiation were also differentially expressed in the DLBCL cases, and were independent of other expression signatures, suggesting that they could be used to define different subsets [Alizadeh et al. 2000; Wiestner and Staudt, 2003]. Genes associated with GCB DLBCL included known markers of germinal center differentiation such as CD10. They also included the bcl-6 gene, which may be translocated or mutated in DLBCL, as well as numerous new genes (Figure 2A) [Dalla-Favera et al. 1999]. In contrast, most genes that defined ABC DLBCL were not expressed by normal GC B cells, but instead were induced during in vitro activation of peripheral B cells such as cyclin D2 and CD44. The ABC DLBCL signature also included the IRF4 (MUM1) gene that is necessary for antigen receptor driven B-cell proliferation [Matsuyama et al. 1995; Mittrucker et al. 1997]. A noteworthy feature of ABC DLBCL was the expression of bcl-2 that is induced over 30-fold during peripheral B-cell activation [Tschopp et al. 1998]. Most ABC DLBCLs had over fourfold higher bcl-2 expression compared with GCB DLBCLs [Alizadeh et al. 2000].

Diagnosis and outcome of diffuse large B-cell lymphoma (DLBCL) subtypes by gene expression profiling subtypes. (A) Heat map showing expression of genes that discriminate between the germinal center B-cell like subtype (GCB) and activated B-cell like subtype (ABC) subtypes of DLBCL. Genes associated with the microenvironment, which have prognostic significance, are clustered into stromal 1 and 2 signatures. The stromal 1 signature genes are associated with extracellular matrix deposition and histiocytic infiltration and the stromal 2 signature genes are associated with increased tumor blood vessel density. (B) Kaplan–Meier estimates of progression free and overall survival are shown according to GCB or ABC DLBCL subtype in patients treated with R-CHOP based therapy. Median follow up is approximately 2 years [Lenz et al. 2008]. (Image courtesy of Dr Louis Staudt, NCI.)
These results suggested that the GCB and ABC DLBCL subtypes are derived from B cells at different stages of differentiation. GCB DLBCL appears to arise from GC B cells whereas ABC DLBCL likely arises from post-GC B cells that are blocked during plasmacytic differentiation. Genetic analysis has revealed ABC and GCB DLBCL to be pathogenetically distinct. GCB DLBCL is exclusively associated with two recurrent oncogenic events, the t(14;18) translocation involving the bcl-2 gene and amplification of the c-rel locus on chromosome 2p. They also have amplification of the oncogenic mir-17-92 microRNA cluster and deletion of the tumor suppressor PTEN. In addition, frequent abnormalities of Bcl-6 are found [Lenz et al. 2008b; Parekh et al. 2007, 2008; Shaffer et al. 2002].
ABC DLBCLs have frequent amplification of the oncogene SPIB, deletion of the INK4a/ARF tumor suppressor locus and trisomy 3. The NF-kB pathway is constitutively activated in most ABC DLBCL cases [Davis et al. 2001; Lenz et al. 2008a; Ngo et al. 2006]. This has been linked to abnormalities in a variety of upstream proteins, including CARD11, BCL10 and A20 leading to activation of IκB kinase and NF-κB activation [Davis et al. 2001; Lenz et al. 2008a; Ngo et al. 2006]. For example, 10% of ABC DLBCL cases have somatic mutations in CARD11, a signaling scaffold protein, that cause it to constitutively engage the NF-κB pathway (Figure 3) [Lenz et al. 2008a]. For the majority of ABC DLBCL cases, NF-κB activation can be observed in the absence of CARD11 or BCL10 mutations. Until recently, alternative mechanisms of NF-κB activation were poorly understood. However, recent work has demonstrated that NF-κB activation may be linked to chronic active B-cell receptor (BCR) signaling. Targeting the BCR pathway component, Bruton’s tyrosine kinase (Btk), resulted in a significant in vitro antiproliferative activity against ABC but not GCB DLBCL [Davis et al. 2010]. To provide genetic evidence of BCR signaling in the pathogenesis of ABC DLBCL, genes in the BCR pathway in DLBCL cell lines and biopsies were sequenced [Davis et al. 2010]. Missense mutations in CD79B protein of the BCR were identified in two cell lines, and in 21% of ABC DLBCL and 3% of GCB DLBCL tumor biopsies [Davis et al. 2010]. These results suggest that a significant proportion of ABC DLBCL may have a heightened BCR antigenic response, leading to abnormal activation of NF-κB.

B-cell receptor (BCR) signaling pathway and potential targets. (A) Signaling through BCR leads to downstream activation of the nuclear factor kappa B (NF-κB) transcription factor, which is a driver pathway in activated B-cell like subtype (ABC) diffuse large B-cell lymphoma (DLBCL). Signaling also activates the Akt/mTOR and MAP kinase pathways. (B) Targeted therapies in ABC DLBCL to inhibit NF-κB are dependent on the presence or absence of activating mutations in CARD11. Tumors with activating mutations are likely to require inhibition of downstream targets (e.g. NF-κB activation) whereas those with wild type are likely to be sensitive to both downstream and upstream (e.g. inhibition of BTK or SYK) targets. (Image courtesy of Dr Louis Staudt, NCI.)
BCR signaling also activates the PI3K/Akt/mTOR signaling pathway with effects on apoptosis, proliferation and metabolism. A recent study has also shown ABC DLBCL has a dependence on MYD88, an adaptor protein that mediates TOLL and interleukin (IL)-1 receptor signaling [Ngo et al. 2010]. RNAi screening showed that MYD88 and the IL-1 receptor-associated kinases (IRAK), IRAK1 and IRAK4, are indispensable for ABC DLBCL survival. Analysis of ABC DLBCL tumors revealed that 29% harbored mutations in MYD88. The L265P mutant promoted cell survival through assembling an IRAK1 and IRAK4 protein complex, which lead to NF-κB signaling, JAK kinase activation of STAT3, and secretion of IL-6, IL-10 and interferon-β. These results indicate that the MYD88 signaling pathway is central to the pathogenesis of ABC DLBCL, and supports the development of inhibitors of this pathway. These results suggest a number of strategies to exploit chronic active BCR signaling in ABC DLBCL (Figure 3).
If the molecular taxonomy defines true DLBCL subtypes, it should also have prognostic value. An analysis of molecular subtype and outcome following upfront CHOP treatment demonstrated a statistically significant difference in overall survival at 5-years of 59% in GCB and 31% in ABC subtypes of DLBCL, and these were independent of the IPI risk groups [Alizadeh et al. 2000; Wiestner and Staudt, 2003]. Because this analysis was performed on biopsies obtained in the prerituximab era, a second analysis was performed on 233 biopsies obtained from patients treated with R-CHOP [Lenz et al. 2008b]. Similar to the aforementioned results, patients with GCB compared with ABC DLBCL had a more favorable survival, with 3-year overall survival rates of 84% versus 56%, respectively (p < 0.001); expectedly, both GCB and ABC DLBCL performed better compared with the prerituximab analysis (Figure 2B). In this analysis, a new ‘stromal 2’ signature was discovered that predicted inferior survival, whereas a stromal 1 signature was found to be favorable (Figure 2A). The stromal 1 signature was associated with extracellular matrix deposition and histiocytic infiltration and the stromal 2 signature associated with increased tumor blood vessel density. Although the stromal 2 signature suggests that anti-angiogenesis treatment may be useful, it is also possible that this signature is secondary to an anaerobic environment and is not a driver event.
It is important to note that the molecular distinctions between the GCB and ABC DLBCL subtypes have yet to have clinical application. However, they are critically important to advance the targeted treatment of DLBCL. In this regard, the best practical method(s) for identifying these subtypes remains a matter of controversy. While GEP on frozen tissue remains the ‘gold standard’, it has obvious practical limitations for clinical practice. In its place, investigators have developed immunohistochemical predictor models, which have had variable reproducibility, but nonetheless have successfully distinguished GCB from non-GCB DLBCL in a number of clinical trials [Choi et al. 2009; Dunleavy et al. 2004; Gutierrez-Garcia et al. 2011; Hans et al. 2003]. Recent advances in paraffin-based GEP will likely emerge as the new standard due to its ability to replicate the validated GEP expression signatures for GCB and ABC DLBCL.
Targeting GCB DLBCL
While GCB DLBCL has a better prognosis than ABC DLBCL, upwards of 30% of patients are not cured with initial immunochemotherapy. The resistance of GCB DLBCL to curative treatment may relate to the effect of Bcl-6 on cell growth and survival [Phan and Dalla-Favera, 2004; Phan et al. 2005]. Bcl-6 is an important modulator of B-cell development in the GC, and its transcriptional silencing is required for exit of the B-cell from the GC. Bcl-6 suppresses genes that are involved in lymphocyte activation, differentiation, cell cycle arrest, and include p21 and p27Kip1, and the DNA damage response genes, p53 and ATR [Phan and Dalla-Favera, 2004]. In GCB DLBCL, chromosomal translocations affecting the Bcl-6 locus juxtapose heterologous promoters from the partner chromosome with intact Bcl-6 coding sequences, leading to deregulated expression of Bcl-6; in addition, Bcl-6 can be altered by multiple somatic mutations. These mutations/translocations in Bcl-6 enhance its inhibitory effect on the apoptotic stress response and promote proliferation, both of which are associated with treatment failure [Paik et al. 2005; Pasqualucci et al. 2003; Phan and Dalla-Favera, 2004; Ranuncolo et al. 2007; Wilson et al. 1997].
These results suggest that Bcl-6 is an important target for GCB DLBCL. Inhibitors targeting the Bcl-6 BTB domain protein interaction have shown efficacy in vitro [Cerchietti et al. 2010; Parekh et al. 2008]. Targeting other Bcl-6 domains or using histone deacetylase inhibitors to overcome Bcl-6 repression of p53 and cell cycle inhibitory proteins may also be potentially useful, and are under investigation [Parekh et al. 2008]. An interesting and potentially important observation is the effect of topoisomerase II inhibition on Bcl-6 levels. It has been shown that the topoisomerase II inhibitor etoposide leads to downregulation of Bcl-6 expression by ubiquitin-mediated protein degradation and possibly through transcriptional inhibition [Kurosu et al. 2003]. This may partially account for the in vitro finding that sustained exposure of tumor cells to etoposide and low-dose doxorubicin promote the p53–p21 pathway and activates the check-point kinase (Chk2), effects that are inhibited in cells engineered to overexpress Bcl-6 [Siu et al. 2004; Theard et al. 2001]. This raises the possibility that inhibition of topoisomerase II may be particularly important in GCB DLBCL. In this regard, the German High-Grade Lymphoma Study Group showed that the addition of etoposide to CHOP (CHOEP) significantly improved the event-free survival of younger but not older patients with untreated DLBCL [Pfreundschuh et al. 2004a, 2004b]. The higher frequency of GCB DLBCL in younger compared with older patients may explain why the benefit of etoposide was only found in the study of patients under 60 years and not over 60 years [Pfreundschuh et al. 2004a, 2004b; Rosenwald et al. 2002]. Interestingly, the positive effect of including etoposide in CHOEP was lost when rituximab was added (R-CHOPE) [Pfreundschuh et al. 2006]. However, this may reflect the overall salutary effect of rituximab on the outcome of DLBCL, including both GCB and ABC DLBCL, and not a specific effect on Bcl-6.
The association between topoisomerase II inhibition and inhibition of Bcl-6 raises the hypothesis of whether regimens that highly inhibit topoisomerase II would be more effective in GCB DLBCL, even in the setting of rituximab. Interestingly, the DA-EPOCH-(R) regimen, which was designed to inhibit topoisomerase II through several strategies, has demonstrated very high efficacy in GCB DLBCL across several studies and in a recent multi-institutional study, patients with GCB DLBCL achieved a 100% event-free survival at a median follow-up time of 5 years [Wilson et al. 2002, 2011; Dunleavy et al. 2010].
Targeting ABC DLBCL
As discussed earlier, studies have demonstrated that ABC DLBCLs, which are associated with an inferior clinical outcome, are characterized by constitutive activity of NF-κB, which activates genes associated with tumor cell survival and proliferation. To help assess whether NF-κB was a clinically useful target, we undertook a ‘proof of principle’ clinical study to test whether inhibition of NF-κB might sensitize ABC but not GCB DLBCL to chemotherapy (Figure 4A and B) [Orlowski and Baldwin, 2002; Orlowski and Kuhn, 2008]. Based on in vitro evidence that bortezomib, a proteasome inhibitor, blocked degradation of phosphorylated IκBα and consequently inhibited NF-κB activity in ABC DLBCL cell lines, bortezomib was combined with DA-EPOCH in patients with relapsed/refractory DLBCL [Allen et al. 2008; Houldsworth et al. 2008; Strauss et al. 2007]. Tumor tissue was analyzed by GEP and/or immunohistochemistry to identify molecular DLBCL subtypes (Figure 4A). As a control, it was demonstrated that relapsed/refractory ABC and GCB DLBCL have equally poor survivals following upfront chemotherapy. Bortezomib alone had no activity in DLBCL, but when combined with chemotherapy, it demonstrated a significantly higher response (83% versus 13%; p = 0.0004) and median overall survival (10.8 versus 3.4 months; p = 0.0026) in ABC compared with GCB DLBCL, respectively (Figure 4B). These results suggest that bortezomib enhances the activity of chemotherapy in ABC but not GCB DLBCL, and provide a rational therapeutic approach based on genetically distinct DLBCL subtypes [Dunleavy et al. 2009]. In another recent study, bortezomib was combined with R-CHOP in patients with previously untreated DLBCL to assess its toxicity and differential efficacy in molecular subtypes [Ruan et al. 2010]. In this study of 40 patients with DLBCL, the progression-free survival was 64% at 2-years and there was no difference among patients with GCB and ABC DLBCL. One would expect an inferior outcome for patients with the ABC subtype so these results suggest that bortezomib overcame the adverse prognostic effect of the ABC DLBCL subtype. Based on these studies, a randomized study of R-CHOP ± bortezomib in untreated patients with ABC DLBCL is ongoing (PYRAMID Study).

Clinical treatment paradigm. (A) Patients initially received bortezomib alone at 1.3 mg/m2 on days 1, 4, 8 and 11 every 21 days (Part A) unless they had disease which the investigators judged to required immediate chemotherapy such as impending or ongoing organ compromise; these patients only received Part B. Patients with progressive disease on Part A received bortezomib with DA-EPOCH (Part B). Of 31 diffuse large B-cell lymphoma (DLBCL) cases analyzed by gene expression profiling, 16 were excluded due to ineligible subtype by classification or did not receive Part A, leaving 5 activated B-cell like subtype (ABC) and 10 germinal center B-cell like subtype (GCB) cases eligible for analysis of outcome. Of 24 paraffin-embedded tumor biopsies analyzed by immunohistochemistry, 12 each were categorized as GCB and ABC (non-GCB) type [Hans et al. 2003]. By combining both methods, cases were identified as GCB in 15 and ABC in 12 and included in the analysis of outcome with Part B. (B) Response and overall survival of 27 patients with de novo GCB or ABC DLBCL who received DA-EPOCH-B. Overall survival of patients with ABC or GCB DLBCL showed a median survival of 10.8 and 3.4 months, respectively (p = 0.0026). Patients with ABC DLBCL also had a significantly higher complete and overall response rate compared with patients with GCB DLBCL [Dunleavy et al. 2009].
While it is important to show that inhibition of NF-κB is clinically useful, it is also important to understand and target upstream driver events, that activate NF-κB. As discussed, chronic BCR signaling, and activating mutations of CARD11 and MYD88 lead to NF-κB activation (Figure 3B). These results suggest a number of strategies to exploit chronic active BCR signaling in ABC DLBCL. One such target is Btk, where a selective Btk inhibitor, ibrutinib (PCI-32765), was shown to be selectively toxic to cell lines with chronic active BCR signaling (and a clinical study of this agent in DLBCL is underway [Staudt et al. 2011]) [Davis et al. 2010]. Importantly, the position of molecular lesions in the BCR and NF-κB signaling pathways could help guide therapy of ABC DLBCL. For example, ABC DLBCLs with wild-type CARD11 and chronic active BCR signaling might respond to a Btk inhibitor, and possibly to inhibitors of Src-family kinases, PKC-β or Syk, in some cases (Figure 3B). (A study of the Syk inhibitor, fostamatinib disodium, has been done in relapsed non-Hodgkin lymphoma and demonstrated some clinical activity [Friedberg et al. 2010]). In contrast, CARD11-mutant tumors would require agents that target downstream components of the NF-kB pathway. A precise assessment of which ABC DLBCL cases depend on chronic active BCR signaling will require the development of predictive biomarkers and the results of clinical trials involving BCR signaling inhibitors, such as Btk.
Based on these observations and insights, a pilot study of the Btk inhibitor, PCI-32765 is ongoing in patients with relapsed/refractory ABC DLBCL. Early results of this study demonstrated activity of PCI-32765 in relapsed ABC lymphoma and paired mutational analysis in the study showed that the drug modulates chronic active B-cell receptor signaling in responders (in whom CD79B mutations were found) [Staudt et al. 2011].
The Syk inhibitor, fostamatinib, has not been specifically studied in ABC lymphoma but has activity across a wide range of lymphomas including DLBCL [Friedberg et al. 2010]. In one study, response rates were observed in 22% of patients with DLBCL [Friedberg et al. 2010].
There are also studies that have targeted the PI3K/AKT/mTOR signaling pathway using mTOR inhibitors in patients with relapsed Hodgkin and non-Hodgkin lymphomas. Although the patients have been heterogeneous, mTOR inhibitors (temsirolimus and everolimus) have induced complete remissions across lymphoma subtypes [Smith et al. 2010; Witzig et al. 2005]. These results suggest that different types of lymphomas, including DLBCL, are dependent on an activated PI3K/Akt/mTOR pathway, including DLBCL. Although the ideal target for the PI3K/Akt/mTOR pathway is unknown, investigators are targeting upstream molecules such as Akt and PI3K and a recent trial using the PI3K inhibitor CAL-101 yielded responses in a variety of lymphoid malignancies [Flinn et al. 2009]. Thus, inhibitors of mTOR and/or upstream targets such as Akt and PI3K need to be evaluated in ABC DLBCL.
A recent study suggests that lenalidomide, an immune modulatory agent, may also be preferentially effective in ABC DLBCL [Hernandez-Ilizaliturri et al. 2011]. In a 40-patient phase II study of relapsed/refractory DLBCL, lenalidomide produced had a 29% complete and 53% overall response rate in non-GCB (surrogate of ABC DLBCL) compared with 10% in GCB DLBCL. The median overall survival was also significantly longer in non-GCB (187 days) compared with GCB DLBCL (51 days) (p = 0.004). Although the mechanisms of action of lenalidomide are complex and incompletely understood, these investigators demonstrated that lenalidomide inhibits NF-κB in a Raji cell NF-κB activity reporter assay, and also inhibits angiogenesis [Hernandez-Ilizaliturri et al. 2005; Reddy et al. 2008]. Thus, lenalidomide is another important agent that warrants further investigation in ABC DLBCL.
Targeting MYC and BCL2
The studies we have discussed thus far demonstrate that GCB and ABC DLBCL have different driver pathways, derived from normal pathways associated with their cell of origin. However, they also share potential targets that perform differently according to the subtype of DLBCL. One example of this is BCL2, which is expressed in both GCB and ABC DLBCL. While some older studies found an association between BCL2 expression and poor outcome in DLBCL, later studies have shown a more complex association [Dunleavy and Wilson, 2011; Gascoyne et al. 1997; Iqbal et al. 2011; Wilson et al. 1997]. The mechanism of BCL2 ‘overexpression’ has been related to its prognostic relevance in DLBCL. Gascoyne and colleagues showed that BCL2 overexpression was only associated with a poor outcome in the absence of a t(14:18), which indicates that the mechanism of expression and not the protein itself is more relevant to prognosis [Gascoyne et al. 1997]. This becomes more understandable when considering the relationship of bcl-2 expression to the molecular subtype of DLBCL. In GCB DLBCL, BCL2 expression is typically associated with t(14:18), which is only found in GCB DLBCL, whereas in ABC DLBCL, BCL2 overexpression is associated with gene amplification or NF-κB transcriptional activation [Alizadeh et al. 2000; Rosenwald et al. 2002]. In this latter case, BCL2 expression may simply be a surrogate biomarker for ABC DLBCL, and may not in itself be an important therapeutic target. It is interesting that in a recent study where gene expression profiling was used, BCL2 protein overexpression was a biomarker of poor outcome in the GCB group but not in the ABC group and this suggests that there are diverse mechanisms of BCL2 activation [Dunleavy and Wilson, 2011; Iqbal et al. 2011]. It is likely that BCL2 is an important druggable target and agents such as ABT-199 are under development [Wilson et al. 2010b].
Another potentially important biomarker and target is MYC. High MYC expression is of course observed in Burkitt lymphoma but is also observed in DLBCL where it is associated with the proliferation signature [Alizadeh et al. 2000; Rosenwald et al. 2002]. Recent studies have also shown that up to 10% of DLBCL cases harbor MYC translocations, and these are associated with a poor outcome with standard R-CHOP treatment [Klapper et al. 2008; Savage et al. 2009]. Expectedly, MYC translocation was associated with significantly higher tumor proliferation. Furthermore, MYC translocations were present in both GCB and non-GCB (surrogate of ABC) DLBCL and were adverse in both groups. These studies suggest that newly diagnosed patients with DLBCL should have their tumors analyzed for a MYC translocation and receive treatments other than R-CHOP. While the optimal treatment approach is unknown, in one study 108 cases of DLBCL treated with DA-EPOCH-R were probed for MYC translocations and MYC+ and MYC– cases had a similar EFS at 5 years (83% and 76%, respectively) [Dunleavy et al. 2011b].
Molecular pathology of primary mediastinal B-cell lymphoma
GEP has also been applied to primary mediastinal B-cell lymphoma (PMBL), an important subtype of DLBCL that mostly occurs in young patients [Abou-Elella et al. 1999]. This subtype is defined by a combination of clinical and pathological features, which often resemble classical Hodgkin lymphoma and can confound an accurate diagnosis [Gonzalez et al. 1991; Jaffe et al. 2001]. Two recent studies using GEP have confirmed the unique biological identity of PMBL and demonstrated a strong relationship between PMBL and Hodgkin lymphoma (Figure 5A and B) [Rosenwald et al. 2003; Savage et al. 2003]. Cases of PMBL could be accurately identified by a model using 35 genes that were more highly expressed in PMBL and 11 genes that were more highly expressed in DLBCL [Rosenwald et al. 2003]. When this model was applied to 46 patients with a diagnosis of PMBL, 76% were classified as PMBL. Of the remaining 11 cases, however, 7 and 4 were classified as belonging to the GCB and ABC DLBCL subtypes, respectively, indicating that, although these latter cases predominantly involved the mediastinum, they were not PMBL. Clinically, cases identified as PMBL by gene expression appeared to have a relatively favorable 5-year survival of 64% compared with 59% and 30%, respectively, for the GCB and ABC DLBCL subtypes.
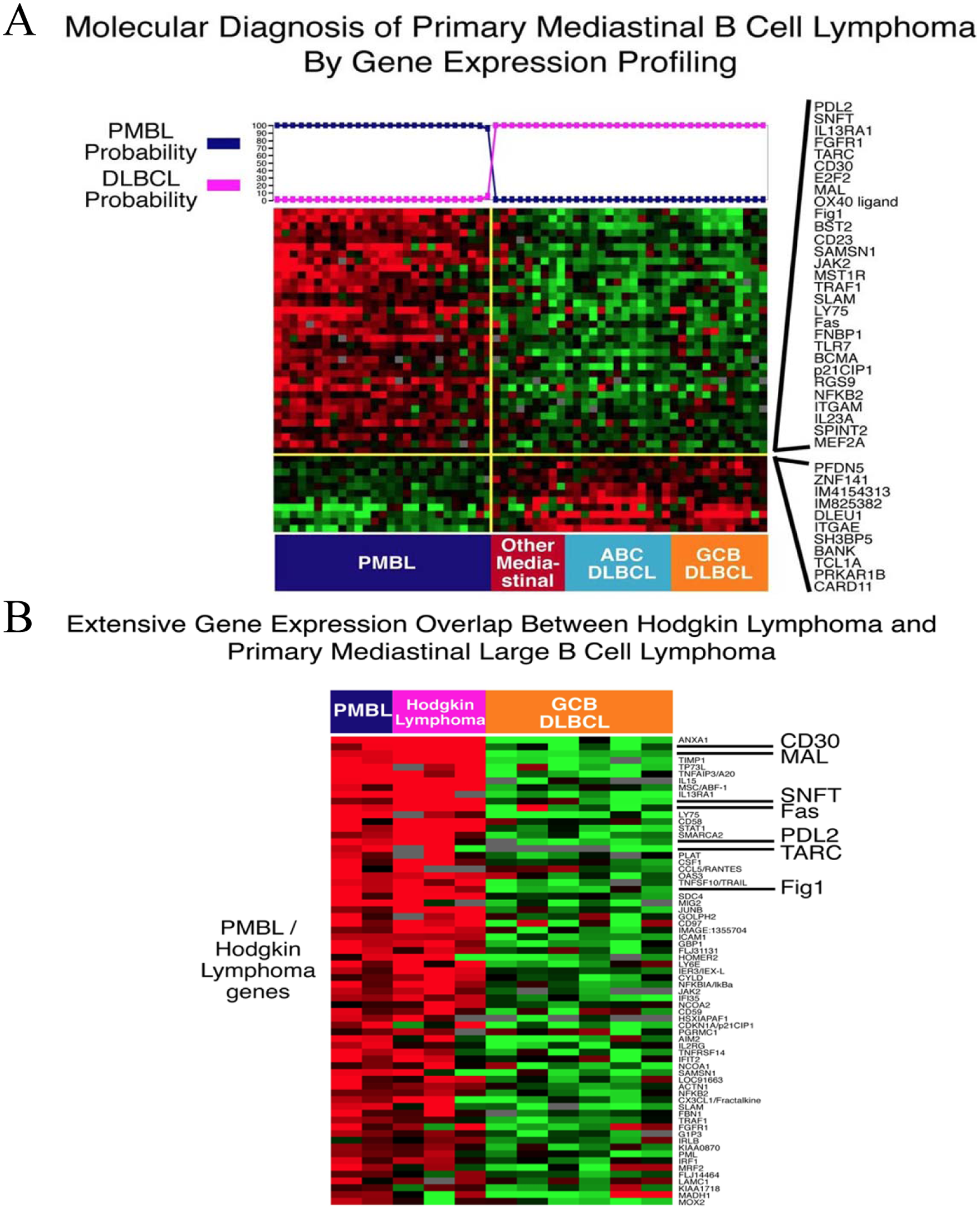
Gene expression profiling of primary mediastinal B-cell lymphoma (PMBL) and comparisons with germinal center B-cell like subtype (GCB) and activated B-cell like subtype (ABC) diffuse large B-cell lymphoma (DLBCL) and Hodgkin’s lymphoma. (A) Heat map of genes that discriminate PMBL from other mediastinal large B-cell lymphomas and GCB and ABC DLBCL. (B) Heat map showing overlap in gene expression between PMBL and Hodgkin’s lymphomas, including CD30 and PDL2 [Rosenwald et al. 2003]. (Image courtesy of Dr Louis Staudt, NCI.)
Over half of PMBL cases and three Hodgkin lymphoma cell lines had gains/amplifications in a region of chromosome 9p. The amplicon on chromosome 9p includes JAK2, which encodes a tyrosine kinase, and SMARCA2, which encodes a putative chromatin regulator. Functional studies are needed to assess the relative contributions of each of these chromosome 9p genes to the pathogenesis of PMBL [Rosenwald et al. 2003]. To identify oncogenes in this amplicon, an RNAi screen was performed targeting amplicon genes and identified JAK2 and the histone demethylase, JMJD2C, as essential genes [Rui et al. 2010]. Inhibition of JAK2 and JMJD2C cooperated in killing these lymphomas by decreasing tyrosine 41 phosphorylation and increasing lysine 9 trimethylation of histone H3, promoting heterochromatin formation. MYC, a major target of JAK2-mediated histone phosphorylation, was silenced after JAK2 and JMJD2C inhibition, with a corresponding increase in repressive chromatin. Thus, JAK2 and JMJD2C cooperate to remodel the PMBL epigenome, and this provides rationale for developing JAK2 and JMJD2C inhibitors in this disease.
Targeting PMBL
For the most part, PMBL is approached therapeutically in a similar way to other subtypes of DLCBL, but with regimens such as R-CHOP followed by involved field radiation there is a high rate of treatment failure and the results are suboptimal [Abramson et al. 2011; Aviles et al. 2012; Savage et al. 2006]. In addition, the long-term consequences of mediastinal radiation in this young population of patients are devastating and therapeutic approaches that obviate the need for radiation while maintaining high cure rates are needed [Castellino et al. 2011; Dunleavy and Bollard, 2011]. Studies have suggested that more dose-intense regimens such as MACOP-B or VACOP-B yield a superior outcome compared with CHOP, raising a question of the optimal chemotherapy for PMBL [O’Reilly et al. 1991; Savage et al. 2006; Todeschini et al. 2004; Zinzani et al. 1999, 2002]. Interestingly, the benefit of dose intensity in PMBL is supported by molecular evidence showing its close biological relationship with nodular sclerosis Hodgkin lymphoma, where the value of dose-intense regimens such as escalated BEACOPP is well demonstrated [Diehl et al. 2003]. Based on such evidence that dose intensity is important in PMBL, DA-EPOCH-R, a dose intense regimen, has been studied without radiotherapy in PMBL [Dunleavy et al. 2006; Wilson et al. 2004]. In a recent update of 40 patients with untreated PMBL, the event-free survival and overall survival were 95% and 100%, respectively, at the median follow up of 4 years. Importantly, only two patients required consolidation radiation treatment and no patients have progressed [Dunleavy et al. 2011a]. These results suggest that DA-EPOCH-R obviates the need for radiation in most patients with PMBL, thus eliminating the risk of long-term radiation-associated toxicities such as secondary malignancies and heart disease. This is particularly important given that patients afflicted with PMBL are typically young and often female with an increased risk of breast cancer.
Although the outcome of PMBL is excellent with regimens such as DA-EPOCH-R, it would be important to further reduce the toxicity and length of treatment. Hence, novel targeted agents should be investigated in this disease. In this regard, RNAi screens have identified JAK2 as a potentially important target for PMBL. Mutations of JAK2 have been implicated in myeloproliferative disorders and the selective JAK 1/2 inhibitor, INCB18424 from Incyte corporation, has shown significant activity in these diseases [Quintas-Cardama et al. 2011; Verstovsek et al. 2010]. Presently trials are planned to assess inhibitors of the JAK pathway in DLBCL including PMBL but no clinical data is available at this time.
Conclusions
Although, for the most part, patients with a diagnosis of DLBCL are treated in a similar manner, we now realize that DLBCL is a heterogeneous disease molecularly and can be divided into at least three distinct subtypes. These subtypes have distinct oncogenic characteristics and mechanisms of activation and it is rational to investigate targeting complexes within individual subtypes. It is emerging that some biomarkers such as BCL2 and MYC may be important across different subtypes and novel ways of targeting these are also being pursued. It is hoped that progress in understanding the molecular pathogenesis of these diseases will pave the way for effective targeted therapies that will hopefully improve the curability of especially ABC DLBCL.
Footnotes
Funding
This work was funded by the intramural program of the National Cancer Institute.