
Abstract
Anaplastic lymphoma kinase (ALK) alterations, including activating mutations, amplifications, and fusions/rearrangements, are found in approximately 3.3% of cancers, including over 50% of inflammatory myofibroblastic tumors. Tyrosine kinase inhibitors to target ALK have significant activity against ALK-mutant cancers, including next generation inhibitors to combat frequent resistance. Here, we present a patient diagnosed with high grade metastatic inflammatory myofibroblastic tumor driven by a RANBP2::ALK fusion, who later developed an ALK G1202R resistance mutation in the setting of treatment with crizotinib. Upon changing therapy to lorlatinib, which is effective against this mutation in lung cancer, the patient again achieved a response that permitted surgical resection. The patient remains without evidence of disease now 18 months after discontinuing adjuvant lorlatinib. This case illustrates the importance of serial molecular profiling to guide selection of the optimal ALK inhibitor for the best clinical outcomes.
Introduction
Inflammatory myofibroblastic tumor (IMT) is an ultra-rare mesenchymal tumor with an incidence of less than one per million people. IMTs predominantly affect children, adolescents, and young adults, though they can occur at any age. 1 While these tumors can be indolent, a subset of epithelioid-type IMTs are aggressive with metastatic potential, termed epithelioid inflammatory myofibroblastic sarcoma (EIMS),2,3 which may have limited or short-lived responses to traditional chemotherapy. 4 Importantly, approximately 50% of IMTs, including most EIMS express anaplastic lymphoma kinase (ALK) and are driven by ALK fusions/rearrangements that result in a chimeric fusion protein 2,3,5–7. Since ALK fusions also occur in other cancers, including anaplastic large cell lymphomas (ALCLs) and 3.1% of non-small cell lung cancers (NSCLC), ALK inhibitors may serve as a tumor-agnostic therapy for this target, including later generation agents designed to address frequent emergence of resistance.8–10 Whereas only crizotinib, a first generation ALK inhibitor, has been approved for ALK-aberrant IMTs in adult and pediatric patients, 11 there are four later-generation ALK inhibitors targeted at resistance mutations that have been approved for NSCLC, including: alectinib, brigatinib, ceritinib, and lorlatinib. 9
Here, we report a case of an adult patient with metastatic EIMS bearing an RANBP2::ALK fusion who underwent selection of targeted therapy guided by emergence of the resistance mutation ALK G1202R and achieved durable resolution of disease. The case is presented in accordance with the CARE case report guidelines, Supplementary Data.
Case description
A 34-year-old female patient with no significant past medical history presented to her primary care physician in August 2019 with fatigue, weight loss, night sweats, fevers, and body aches. She was presumed to have infection with unknown source and prescribed azithromycin. In November 2019, the patient presented to her local ER after awakening from her sleep with right lower quadrant pain that was reminiscent of a prior ovarian cyst rupture. Laboratory workup was notable for elevated C reactive protein (CRP) of 99.6 mg/L (normal range 0 – 2.99 mg/L), mild anemia with hemoglobin of 11.6, and elevated platelets at 584 × 10^3. Albumin was reduced at 2.7 with normal electrolytes, renal and hepatic function. Transvaginal ultrasound was concerning for right ovarian torsion; thus she underwent emergent diagnostic laparoscopy of her abdomen which revealed a large pelvic mass attached to the omentum. She was converted to an open laparotomy and underwent omentectomy revealing multiple masses in the omentum with similar nodules present on the external surface of the transverse colon. On staging imaging, no distant metastatic disease was identified. Pathology revealed epithelioid inflammatory myofibroblastic sarcoma (EIMS) with neoplastic cells having large vesicular nuclei with prominent nucleoli, abundant eosinophilic cytoplasm and were epithelioid to spindled in shape, Figure 1(a). Immunohistochemical (IHC) staining revealed diffuse nuclear membrane expression for ALK, Figure 1(b). IHC also confirmed CD30 expression by tumor cells. Multiplex fusion analysis by targeted next-generation sequencing (NGS) confirmed the presence of an RANBP2::ALK fusion (RANBP2 exon 18 to ALK exon 20). Histologic features of epithelioid inflammatory myofibroblastic sarcoma. (a) Immunohistochemistry with H&E staining of initial resection revealing epithelioid inflammatory myofibroblastic sarcoma (EIMS). Tumor cells are epithelioid to spindled in shape, with abundant eosinophilic cytoplasm, and exhibit large vesicular nuclei with prominent nucleoli. (b) Immunohistochemistry for ALK showing diffuse membrane expression.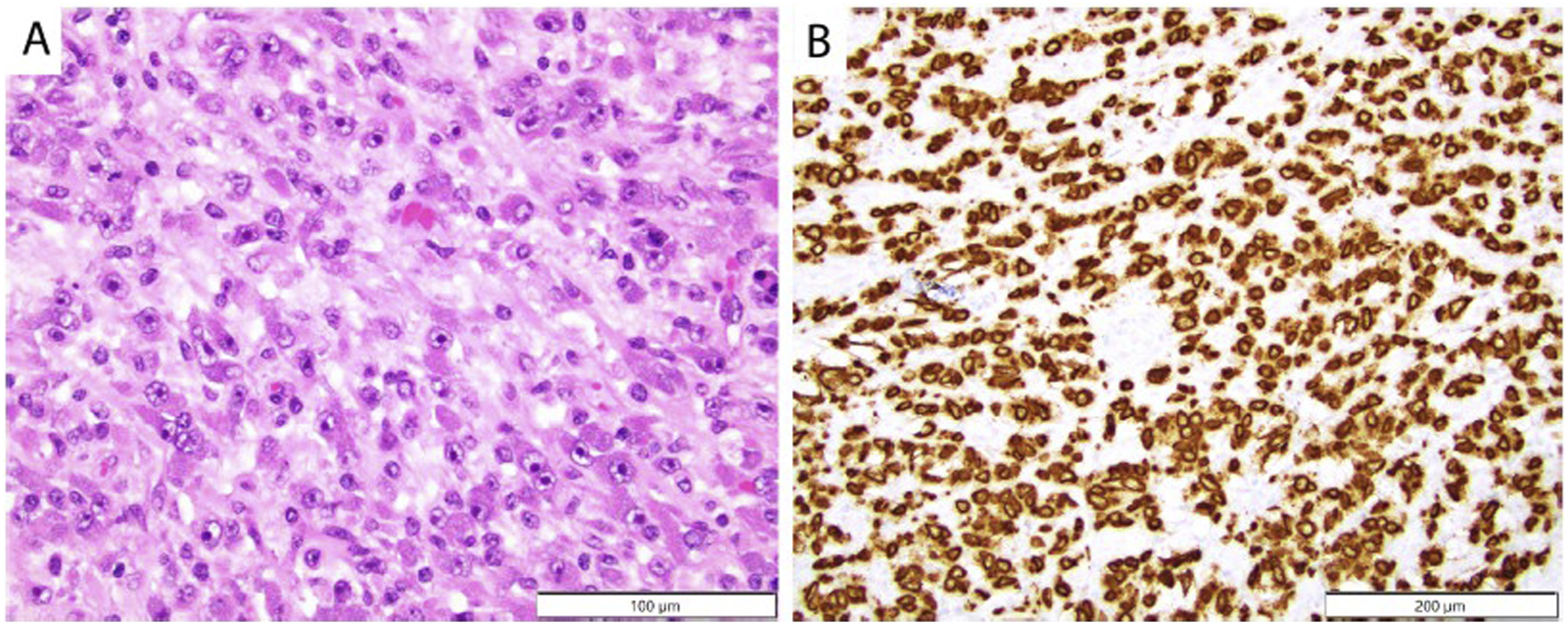
The patient was referred to our center for treatment recommendations. Since her surgery, her fatigue and fevers had improved, but she noted recurrent decreased appetite and suprapubic pain. She underwent a restaging CT of the chest/abdomen/pelvis at our evaluation on December 16, 2019, which revealed recurrent/residual multifocal disease in the peritoneal cavity. A follow up PET/CT scan confirmed extensive peritoneal metastatic disease which was intensely fluorodeoxyglucose (FDG) avid, with no distant metastatic sites, Figure 2(a). Nodular disease was seen throughout the abdomen and pelvis including perihepatic and perisplenic regions as well as the omentum. The patient was initiated on the ALK inhibitor crizotinib 250 mg twice daily on December 22, 2019 with excellent tolerance. Follow up PET/CT after 2 months of therapy revealed a complete response by PET/CT, Figure 2(b). Radiographic imaging over clinical course. (a) PET/CT scan on December 9, 2019 confirmed extensive peritoneal metastatic disease which was intensely fluorodeoxyglucose (FDG) avid. Arrows highlight avid disease (black signal) throughout the peritoneum. (b) Follow up PET/CT after 2 months after initiation of crizotinib revealed resolution of avid disease consistent with a metabolic complete response. (c) PET/CT on August 11, 2020, revealed progression of disease on crizotinib. Arrows depict a large, highly PET avid mass on the transverse colon and additional omental nodules. Lorlatinib was initiated on September 4, 2020. (d) CT imaging on October 21, 2020, revealed marked response in largest lesion on the transverse colon.
About 6 months after initiation of crizotinib, the patient again developed recurrent night sweats and low-grade fevers resembling her initial presenting symptoms. CT imaging in June 2020 showed some mild thickening of the posterior pleura and no definite recurrent lesions. However, a short interval follow up PET/CT obtained August 2020 showed progression of disease with interim development of multiple peritoneal nodules, Figure 2(c). Tumor markers were obtained at that time with elevated CA 27.29 of 56 (normal range <39 U/mL), and CA125 of 124 (normal range 0-35 U/mL). In consultation with our thoracic oncology colleagues, the patient underwent a repeat biopsy of her disease for mutational testing. She was found to have a mutation in the kinase domain of ALK, resulting in an anticipated single amino acid substitution (p.G1202R). In the context of a known ALK fusion-positive tumor with prior treatment with ALK-directed targeted therapy, this finding has been described as a relatively common mechanism of resistance. 9 This mutation is also associated with resistance to second generation ALK inhibitors such as ceritinib and alectinib. No other potential mechanisms of resistance, including additional fusions, gene mutations, or copy number changes were observed. Crizotinib was discontinued September 2020, and the patient was started on the third-generation ALK inhibitor lorlatinib predicted to be effective against the ALK G1202R mutation. 9 Given that lorlatinib is not formally approved for IMT, the patient provided informed consent after a comprehensive discussion of the off-label use, potential risks, benefits, and alternative options. The treatment was reviewed and supported by the institutional tumor board and multiple expert oncologists, in accordance with institutional guidelines for off-label therapies. Although the full dose licensed for NSCLC is 100 mg QD, we elected to start at 50 mg initially for better tolerance.
With treatment, she rapidly demonstrated clinical improvement with resolution of B symptoms, normalization of tumor markers, and an excellent partial response by CT imaging with resolution of all but the largest mass, Figure 2(d). Lorlatinib was increased to 75 mg daily but ultimately decreased back to 50 mg daily due to side effects including arthralgias, fatigue, and elevated lipids. The patient then underwent resection in November 2020 of the residual mass. There were no adhesions or other intraoperative challenges resulting from lorlatinib therapy. Intraoperatively, the only grossly visible disease was the 2 cm mass associated with the transverse colon, and several 1 to 2 mm nodules in the omentum. All visible nodules were removed, and the surgery was considered optimal tumor debulking. On pathology review, >99% of the colon nodule and all of the omental nodules resected showed sequelae of chronic inflammation and marked treatment effect, including foamy histiocytes, foci of fibrosis, and hemosiderin deposition, with <1% remaining viable tumor cells in the colon mass which were positive for perinuclear ALK expression.
Given that there is limited data on adjuvant therapy in this disease following resection, through shared decision making, the patient continued lorlatinib as adjuvant treatment until December 2023, 3 years after her resection. Overall she tolerated therapy reasonably well, with primary toxicities including myalgias, fatigue, appetite decrease, hyperlipidemia, and some subjective cognitive changes including memory difficulties, slowing of executive function, and mood swings. She has remained free of disease without recurrence by symptoms, laboratory markers including CRP, CA27.29, and CA125, or imaging. At the patient’s request, we also monitored Signatera circulating tumor DNA testing beginning March 2022 during her adjuvant therapy with each follow up, and this has remained undetectable since that time, though no baseline test was obtained when she had active disease. Scans and the above laboratory tests were obtained every 3-4 months during adjuvant treatment, and every 6 months post discontinuation of treatment. She is now 18 months from discontinuation of lorlatinib without recurrence of disease and has had resolution of all treatment-related toxicities, Figure 3. Given the aggressive biology of her disease, the fact that she has well exceeded the time to her prior recurrence, and near complete pathologic response with treatment at the time of surgery, her prognosis appears favorable for cure. Timeline of clinical course and treatment. The patient initially presented with B symptoms and abdominal pain in August 2019. She then underwent her initial diagnostic resection in November 2019. Restaging scans postoperatively in December 2019 showed residual/recurrent disease and the patient was initiated on crizotinib. She obtained a complete metabolic response by February 2020. In August 2020, the patient had recurrent disease by PET imaging and biopsy and sequencing confirmed emergence of the ALK G1202R resistance mutation. She was then initiated on lorlatinib and obtained a partial response. She underwent resection in November 2020 with near complete pathologic response of residual lesions. She was continued on lorlatinib as adjuvant therapy until December 2023 and has remained without evidence of recurrent disease to date.
Discussion
This case illustrates a particularly aggressive form of IMT in an adult patient, driven by a RANBP2::ALK fusion, a variant commonly associated with epithelioid inflammatory myofibroblastic sarcoma (EIMS) — an aggressive subtype of IMT known for poor prognosis and high metastatic potential.2–4 Our patient demonstrated an initial robust response to crizotinib, a first-generation ALK inhibitor which was recently approved by the FDA for adult and pediatric patients with unresectable/recurrent ALK-positive IMT, based on a 66.7% response rate and median progression-free survival (PFS) of 18.0 months. 11 However, as with many targeted therapies, acquired resistance emerged, manifesting as disease recurrence within 6 months of crizotinib initiation. Molecular re-evaluation revealed the ALK G1202R mutation, a well-characterized gatekeeper mutation in NSCLC that alters the kinase domain, impeding drug binding and reactivating ALK signaling. 9 In NSCLC, sequential exposure to first- and second-generation ALK inhibitors often leads to the emergence of such resistant clones, with third-generation inhibitors like lorlatinib demonstrating robust activity against these mutations.9,12 Additionally, lorlatinib was superior to crizotinib in the first-line setting for NSCLC, with a 12 months PFS of 78% with lorlatinib compared to 39% with crizotinib. 13
Although the literature on ALK inhibitor resistance and sequencing in IMT/EIMS is limited, several case reports describe promising efficacy of lorlatinib. Lorlatinib was efficacious as upfront neoadjuvant therapy in 2 cases of epithelioid/spindle IMTs bearing EML4::ALK fusions, with no documented progressions while maintained on adjuvant therapy.14,15 Other case reports document response to lorlatinib after failure of entrectinib (TPM4::ALK fusion), 16 after failure of crizotinib (FN-1::ALK fusion, no identified resistance mutation), 17 and after failure of crizotinib and alectinib, with prolonged stable disease of greater than 4 years (ALK rearrangement by FISH, partner unknown). 5 Another case report describes sequential use of ALK TKIs (crizotinib, alectinib, ceritinib and lorlatinib) for a patient with EIMS (PRRC2B::ALK fusion) who developed ALK resistance mutations R1192P on crizotinib, and L1196M on alectinib, with overall survival greater than 2 years. 18
Despite these promising outcomes, some evidence suggests that differences in tumor type and underlying initial ALK fusion partners may affect responses to ALK inhibitors. While the resistance mutation ALK G1202R identified in our patient is commonly reported in NSCLC, very few reports have described this mutation in IMT. Importantly, one patient (EML4::ALK fusion) demonstrated sensitivity to lorlatinib in the setting of an ALK I1171N mutation occurring while on prior alectinib, but subsequently developed an ALK G1202R mutation associated with progression of disease on lorlatinib. 19 Another patient (ALK rearrangement by NGS, partner unknown) also showed lack of response to lorlatinib in the setting of a ALK G1202R resistance mutation occurring on first line alectinib therapy. 20 Although ALK fusions are a hallmark of many IMTs/EIMS, the spectrum of fusions reported differs from those observed in NSCLC. In IMT, ALK rearrangements are detected in approximately 50–60% of cases, with RANBP2::ALK and TPM3/4::ALK among the most frequently reported variants, whereas more than 80% of ALK-driven NSCLC exhibit EML4 as the fusion partner.2,6–9,21 Thus, further investigation will be required to understand how to best sequence ALK inhibitors for treatment of ultra-rare IMT/EIMS.
Our case supports that the underlying biology of ALK-driven EIMS favors stepwise acquisition of kinase domain mutations under selective therapeutic pressure. Thus, early identification of resistance mutations through serial molecular profiling may allow for timely therapy adaptation, maximizing the likelihood of sustained disease control. Alternatively, it is possible that earlier use of a later generation ALK inhibitor such as lorlatinib could prevent emergence of resistance mutations occurring in the setting of crizotinib and lead to more favorable outcomes, as reported in first-line therapy of NSCLC. 13 With crizotinib the only approved agent for IMT/EIMS, it is likely that most patients with unresectable IMT/EIMS will continue to receive this agent first, thus it is critical to recognize the potential for resistance and to identify specific ALK mutations such as G1202R that may warrant use of lorlatinib rather than second-generation inhibitors including alectenib or ceritinib.
Despite the promising activity observed here, not all patients will respond to ALK inhibition, and there is a significant population of IMT/EIMS that are ALK-negative, suggesting a need for alternative therapies. Our patient’s tumor also showed strong CD30 positivity, a feature often seen in EIMS, and one that may potentially inform future combination treatment strategies with CD30 targeted therapies such as brentuximab with preclinical data suggesting significant responses in xenograft models. 22 Both ALK positive and ALK negative IMTs also frequently express PD-L1 and exhibit prominent CD8+ T cell infiltration, suggesting a potential role for PD-1/PD-L1 directed checkpoint inhibitors particularly for ALK-negative tumors. 23 Several case reports have described promising clinical activity with PD-L1 blockade.24–26
While our patient’s response to sequential ALK-targeted therapy provides important clinical insights, we acknowledge that this is a single case report and therefore has inherent limitations in generalizability. Individual patient factors, tumor heterogeneity, and the rarity of IMT/EIMS with RANBP2::ALK fusions limit the extent to which these findings can be extrapolated to the broader patient population. Prospective studies and systematic collection of clinical and molecular data are needed to validate these observations, refine ALK inhibitor sequencing strategies, and better characterize resistance patterns across diverse IMT/EIMS cohorts.
In summary, this case underscores several important clinical lessons: (1) (2) (3) (4) (5)
Conclusion
This case highlights a successful example of precision medicine, utilizing sequential ALK-targeted therapy in a patient with epithelioid IMT with a RANBP2::ALK fusion who developed resistance to crizotinib via an ALK G1202R mutation. Larger, systematic studies are necessary to confirm the optimal selection and sequencing of ALK inhibitors in IMT/EIMS and to guide evidence-based treatment strategies for patients with this rare malignancy.
Supplemental Material
Supplemental Material - Durable response with mutation-guided ALK inhibition in a patient with metastatic epithelioid inflammatory myofibroblastic sarcoma: A case report
Supplemental Material for Durable response with mutation-guided ALK inhibition in a patient with metastatic epithelioid inflammatory myofibroblastic sarcoma: A case report by Sumra S. Chaudhry, D. Ross Camidge, Michael R. Clay, and d Breelyn A. Wilky in Rare Tumors
Footnotes
Consent for publication
The patient provided informed consent for her clinical history and images to be published.
Authorship contribution
Sumra Chaudhry: Conceptualization, Data curation, Writing – Original draft preparation, review and editing. D. Ross Camidge: Conceptualization, Data curation, Writing – review and editing. Michael R. Clay: Data curation, Writing – Original draft preparation, review and editing. Breelyn A. Wilky: Conceptualization, Data curation, Writing – Original draft preparation, review and editing, Supervision, Funding Acquisition.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Pathology Shared Resource of the University of Colorado Cancer Center (National Cancer Institute Cancer Center Support Grant P30CA046934).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.