
Abstract
Soft tissue sarcomas account for less than 1% of new cancer diagnoses, approximately one in five of which are liposarcomas. These tumors typically arise in the deep tissues of the proximal extremity or retroperitoneum, with just under 3% presenting as primary intrathoracic neoplasms. We present an exceedingly rare and particularly unique presentation of primary lung liposarcoma which traversed the mediastinum into the contralateral hemithorax. This report highlights the primary characteristics of the disease and underscores the importance of a multidisciplinary approach to its successful treatment.
Keywords
Introduction
In the United States, less than 1% of all index cancer diagnoses are categorized as soft tissue sarcomas. 1 Of these, liposarcomas account for 15%–25% and are the most aggressive lipomatous tumor type, arising from primitive mesenchymal cells most commonly in the deep soft tissues of the proximal extremity or retroperitoneum in adults aged 40–60 years2,3 Although hematogenous spread to the lungs is the mechanism and most common destination for metastasis, primary intrathoracic presentation accounts for less than 3% of all liposarcomas.2,4 Furthermore, primary liposarcoma of the lung is exceedingly rare and represents just 0.2% of lung cancer diagnoses with very few cases reported in existing literature.5,6 Here, we discuss the approach to successful treatment in this unique population highlighting key aspects in multidisciplinary management, surgical planning and technique, as well as consideration of neoadjuvant/adjuvant therapies to minimize the risk of recurrence.
Case Report
A 40-year-old female with no history of cardiopulmonary disease initially presented to an outside emergency department with a chief complaint of acute unilateral flank pain. CT imaging of the abdomen was performed which incidentally revealed a lesion posterior to the cavo-atrial junction near the coronary sinus. It measured 1.7 × 2.7 × 3.4 cm with a homogenous composition and was marginally denser than the surrounding adipose tissue. Differentials at the time included lymphadenopathy, complex pericardial cyst, and pericardial fat necrosis, among others. It was thought to have a low likelihood of malignancy and Cardiac MRI was recommended to further characterize it. The patient was lost to follow-up for an extended period. She presented to the emergency department 6 years later with recurrent flank pain. CT abdomen performed at this time demonstrated progressive enlargement of the mass but outpatient follow-up was again not performed. She was then evaluated by a primary care physician at this institution 2 years later. Chief complaint at that time included new onset dyspnea on exertion, cough, and finger swelling. Physical exam revealed upper and lower extremity clubbing prompting repeat CT imaging of the chest and referral to pulmonology. The mass had continued to grow and measured 13 cm × 13 cm x 12 cm with extension into the posterior mediastinum and left hemithorax. (Figure 1) PET scan demonstrated rim-enhancement and a CT-guided core needle biopsy revealed large anaplastic cells with multinucleated hyperchromatic nuclei which stained diffusely positive for MDM2 and negative for AE1/AE3, p40, EMA, desmin, SMA, S-100, melanoma cocktail, CD 3/20/30/45, and SOX-10. (Figure 2) Further analysis revealed MDM2 amplification in 72% of cells by FISH testing. Results were consistent with dedifferentiated liposarcoma. She was referred to medical oncology and thoracic surgery for evaluation for potential resection. Cardiac MRI demonstrated abutment of both atria but no direct invasion of the heart, inferior vena cava, or posterior wall of the left ventricle. The case was discussed in a multidisciplinary thoracic tumor board and the recommendation was made to treat the patient with neoadjuvant radiation prior to repeat imaging and evaluation for surgical resection. Axial and coronal CT Chest prior to (a&b) and following (c&d) neoadjuvant radiation therapy. Hematoxylin and eosin (HE) and MDM2 staining of the initial biopsy (a&b), and HE staining of the surgically resected tumor (c).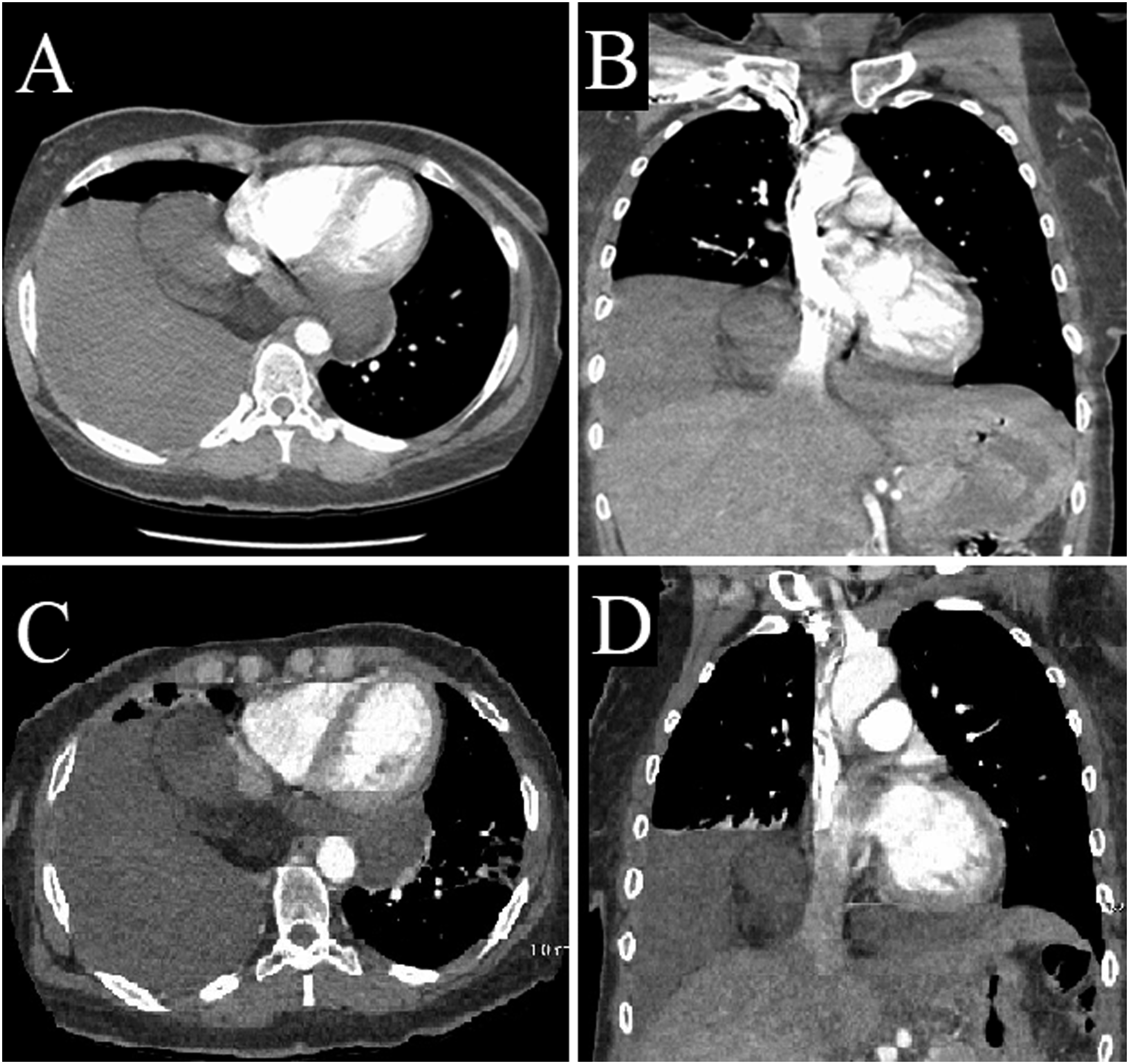

Radiation therapy was delivered to the mediastinum and right lower lobe using intensity modulated radiation therapy (IMRT) at a total dose of 6600 CGY over 30 fractions. CT imaging after completion revealed a slight decrease in the size of the mediastinal portion of the tumor, but enlargement of the portion in the right lower lobe. (Figure 1) Given the reduction in size of the mediastinal component, the decision was made to proceed with surgical resection. Esophagogastroduodenoscopy was performed after induction of anesthesia and confirmed the absence of esophageal invasion or signs of external compression. Initial assessment was performed through right video-assisted thoracoscopy, which was then followed by elective conversion to posterolateral thoracotomy. The 5th rib and intercostal muscles were sent for pathologic analysis upon entry. There was an adhesion between the mass and posterior chest wall, but the mass was otherwise untethered. The SVC and phrenic nerve were identified and used as landmarks to dissect the anterior hilum. The lower lobe vasculature was isolated. Right lower lobectomy was performed following careful release of adhesions and resection of a portion of the pericardium posterior to the phrenic nerve. The mediastinal component of the mass was free of adhesions and was mobilized into the right chest without significant difficulty. The entirety of the mass was removed en-bloc with adequate margin and the operation was completed without issue. (Figure 3) Final surgical pathology was ypT4 according to AJCC 8th edition staging. The mass measured 29.5 × 14.5 × 6.0 cm and was noted to have a 6-cm central focus with near complete necrosis, which likely represented the dedifferentiated component of the neoplasm. The remainder of the tumor demonstrated well-differentiated liposarcoma. (Figure 2) All margins, including adhesions to lung parenchyma, visceral pleura, right chest wall, and pericardium were negative for malignancy. A single subcarinal lymph node was also negative for sarcoma. The patient recovered quickly postoperatively and was discharged home on postoperative day 3. Adjuvant radiation or chemotherapy was not indicated. Surveillance was conducted via CT imaging of the chest, abdomen, and pelvis every 3 months for 1 year and every 6 months in the subsequent 2 years. At 3- year follow-up, the patient is subjectively doing well with no evidence of recurrent or distant disease. Surgical specimen. Green star marking right lower lobe parenchyma, yellow circle marking transmediastinal segment of tumor, purple square marking portion of tumor in the right hemithorax.
Discussion
The World Health Organization identifies five distinct subtypes of liposarcoma: well-differentiated, de-differentiated, myxoid, pleomorphic, and myxoid pleomorphic. 7 Well-differentiated liposarcomas (WDL) are the most common and can be distinguished from lipomas by the presence of hyperchromatic stromal cells, nuclear atypia, and CDK4 and MDM2 amplification on FISH. 8 Though inherently non-invasive, approximately 10%–20% acquire additional complex amplifications sufficient for histological progression after an average of 7-8 years transforming them to the dedifferentiated subtype, which does possess metastatic potential. 3 Dedifferentiated liposarcomas (DDL) arise due to abrupt transformation from WDL. 8 They tend to be particularly aggressive and confer the worst prognosis of primary intrathoracic liposarcoma subtypes when considering disease-free and overall survival, independent of tumor size.2,9 Another subtype, termed myxoid, exists on a spectrum from pure myxoid to increasing round cell morphology, which behaves more aggressively. The hallmark of this subtype is a DDIT3 gene rearrangement due specifically to either a t(12;16) (q13;p11) or t(12;22) (q13q11-12) translocation. 8 Pleomorphic liposarcomas make up the least common subtype and are distinguished histologically from undifferentiated pleomorphic sarcomas by the presence of multivacuolated lipoblasts.2,3,8 Rarely, pleomorphic and myxoid features are observed in conjunction histologically. In such instances, these neoplastic cells lack DDIT3 rearrangements and MDM2 amplification, thereby warranting distinction as pleomorphic myxoid liposarcoma. 8
Existing data on intrathoracic and more specifically, pulmonary liposarcomas, is limited due to rarity. There is more available evidence pertaining to liposarcomas originating elsewhere, which is often used to help guide the treatment of intrathoracic liposarcomas. At present, en-bloc surgical resection with wide margins is the mainstay of treatment and is associated with improved overall survival.10,11 Lymphadenectomy is not routinely performed given that metastasis is typically hematogenous. 2 While data is skewed by inconsistencies in the use of neoadjuvant or adjuvant therapies, a larger series of intrathoracic liposarcoma reported median survival is roughly 11 months for de-differentiated tumors; significantly shorter than pleomorphic (13 months), myxoid (27 months), and well-differentiated (60 months). 9 Local recurrence of disease is not uncommon, with re-resection of retroperitoneal liposarcomas only showing benefit in a select group of patients with slow-growing tumors. 10 As such, minimizing the risk of recurrence is paramount. Though tumor size is not known to be a significant prognostic indicator, the potential for histologic transformation, tumor metastasis, or invasion into surrounding vital structures increases with time and can preclude surgical resection. This point becomes extremely relevant when considering intrathoracic tumors, which can grow quite large before symptoms of chest pain, cough, dyspnea, or shortness of breath are present, if at all. 9
Neoadjuvant radiotherapy has been utilized in retroperitoneal liposarcomas to shrink the tumor and enable wider margins, potentially making resection safer and reducing the risk of recurrence. 12 Prospective studies demonstrating survival benefit with the utilization of adjuvant or neoadjuvant chemotherapy for patients with liposarcoma are sparse, 10 and multiple studies have actually documented greater rates of disease progression than partial response to therapies, highlighting the difficulty in treating this evasive neoplasm.13,14 Within the chest, this may in part be due to necessarily conservative radiotherapy attributable to organ-specific dose limitations for vital structures 15 or advanced tumor grade at time of presentation secondary to histological progression that outpaces a more insidious symptom onset. A case report by Ochi et al., however; noted stabilization of disease according to the Response Evaluation Criteria in Solid Tumors with doxorubicin and ifosfamide-based neoadjuvant chemotherapy in conjunction with radiotherapy prior to surgical resection of presumed mediastinal Ewing sarcoma which was diagnosed post-operatively as dedifferentiated liposarcoma. 16 While successful in isolated cases, no definitive or generalized consensus guiding the use of (neo)adjuvant chemo- or radiotherapy exists. Susceptibility appears attributable to anatomic location and histologic subtype, with myxoid and pleomorphic subtypes being most radiosensitive and chemosensitive to agents such as eribulin or trabectedin. Alternatively, dedifferentiated liposarcomas are minimally sensitive to chemotherapeutics and moderately sensitive to radiotherapy, though immune checkpoint inhibitors such as nivolumab or ipilimumab may prove beneficial for patients with the dedifferentiated subtype.10,17 Given the generally poor prognosis of intrathoracic liposarcomas and their variable responsiveness to radiation and currently approved chemotherapeutics, more research is needed to identify best treatment practices.9,14,18 A number of promising clinical trials are underway attempting to tailor therapy based on tumor-specific mutations.17,18 The relatively limited evidence on which to guide treatment for this unique subset of intrathoracic liposarcoma patients only increases the importance of a multidisciplinary approach to management at experienced cancer centers to achieve the best outcomes possible. 19
Conclusion
This case highlights an extremely rare presentation of transmediastinal primary lung liposarcoma which progressed untreated for nearly a decade. Pulmonary liposarcoma is a poorly understood and highly morbid neoplasm. The majority of evidence and clinical decisions are made on data obtained from retroperitoneal liposarcomas. This report highlights the positive impact of a multidisciplinary approach to successful treatment which ultimately allowed for complete en-bloc surgical resection following neoadjuvant radiotherapy and resulted in recurrence-free survival at the time of this submission despite the high-grade, aggressive nature of this malignancy.
Footnotes
Author contributors
T.W.: manuscript drafting; A.K.: manuscript drafting, manuscript revisions; B.D.S. manuscript review, manuscript revisions; L.D.J. manuscript review, J.C. manuscript review; P.L.L.: conceptualization, oversight/supervision, manuscript review.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.