
Abstract
Objective
Osteoarthritis (OA) is the most prevalent joint disease characterized by the degeneration of articular cartilage and the remodeling of its underlying bones, resulting in pain and loss of function in the knees and hips. As far as we know, no curative treatments are available except for the joint replacement. The precise molecular mechanisms which are involved in the degradation of cartilage matrix and development of osteoarthritis are still unclear.
Design
By analyzing RNA-seq data, we found the molecular changes at the transcriptome level such as alternative splicing, gene expression, and molecular pathways in OA knees cartilage.
Results
Expression analysis have identified 457 differential expressed genes including 266 up-regulated genes such as TNFSF15, ST6GALNAC5, TGFBI, ASPM, and TYM, and 191 down-regulated genes such as ADM, JUN, IRE2, PIGA, and MAFF. Gene set enrichment analysis (GSEA) analysis identified down-regulated pathways related to translation, transcription, immunity, PI3K/AKT, and circadian as well as disturbed pathways related to extracellular matrix and collagen. Splicing analysis identified 442 differential alternative splicing events within 284 genes in osteoarthritis, including genes involved in extracellular matrix (ECM) and alternative splicing, and TIA1 was identified as a key regulator of these splicing events.
Conclusions
These findings provide insights into disease etiology, and offer favorable information to support the development of more effective interventions in response to the global clinical challenge of osteoarthritis.
Introduction
Osteoarthritis (OA) affects about 9.6% of men and 18% of women over 60 years of age, makes it the most common joint disease worldwide. 1 OA is a joint disease characterized by the degeneration of articular cartilage and remodeling its underlying bone. Patients with OA often experience asymmetrical joint pain and joint dysfunction in knees and hips. Physical therapy, nonsteroidal anti-inflammatory drugs (NSAIDs) drugs therapy, percutaneous injection drugs, and joint replacement are the mainly methods for OA treatment. 2 To date, however, no curative treatments are available, and joint replacement is still the expected treatment for end-stage osteoarthritis in hips and knees.3,4 Therefore, OA leads to a higher disability rate, severely reduces the patient’s quality of life, and increases the economic burden on families and society.5,6
The pathogenesis of OA is mainly caused by the synthesis-degradation imbalance of articular cartilage, extracellular matrix, and subchondral bone and mainly manifested as (1) hypertrophy and apoptosis of chondrocytes in the affected joints and subsequent cartilage degeneration and reduction of extracellular matrix, (2) joint biomechanical changes and consequent bone hyperplasia and subchondral bone sclerosis, and (3) excessive inflammatory cells activation and synovial hyperplasia and thickening.7 -9 The main risk factors for OA include changes in joint biomechanics caused by congenital diseases, injury, obesity-related increased joint weight-bearing and metabolic abnormalities, advanced age and related muscle atrophy, and joint degeneration.7,10 -12 However, the molecular mechanism of the degeneration of cartilage matrix and progression of the OA are poorly understood. And therefore, it is still lack of efficient treatment for early-stage OA. The developing technologies such as molecular biology and bioinformatic methods are of great significance to study complex problems in OA, including identification of the specific mechanism of occurrence and progression, screening of targeted therapeutic drugs and other therapeutic methods, and generating strategy for disease prevention. 13 Recent investigation into OA shows articular chondrocyte homeostasis could be disrupted by overweight, aging as well as genetic alterations in tumor growth factor (TGF)-β/Smad, Wnt/β-catenin, and Ihh signaling pathways.14 -17 The upregulation of catabolic enzymes like matrix metalloproteinases (MMP)-13 and ADAMTS5 in most mouse models of OA suggest that these enzymes may serve as the potential therapeutic targets in the intervention of the progression of the OA. 18
Recently, with the development of high-throughput sequencing technology, more and more studies have attempted to find the possible and potential mechanism of the pathogenesis and progression of OA by performing multi-omics sequencing.19,20 For example, Fisch et al. performed RNA sequencing on knee cartilage samples from patients with higher OA score and patients with lower OA score, and found many transcription factors and transcription profiles changed in OA cartilage. By using single-cell RNA sequencing (scRNA-seq), new insights into chondrocyte taxonomy and novel markers for cartilage progenitor cells (CPCs) were established in OA, which may present potential clues for effective and functional manipulation of human OA cartilage regeneration. 22 By integrating analysis of miRNA and mRNA sequencing data, Coutinho elucidated a miRNA interactome and related pathways in OA cartilage tissues, which demonstrated that miRNA affects expression of genes in the cartilage. 23 By integrating genetics with transcriptomics and proteomics, Steinberg discovered molecular trait loci of effector genes and drug repurposing in OA at different omics level. 24 These studies on the abnormal changes of gene expression products in OA at different omics levels provide a basis for the development of new intervention methods. Alternative splicing is an important mechanism that affects gene expression and function at the transcriptional level. However, few study have systematically investigated the role of aberrant alterations in alternative splicing in the pathogenesis and progression of OA, and even fewer have investigated the regulatory mechanisms of alternative splicing or the isoforms switching caused by alternative splicing. This study will focus on identifying the abnormal molecular changes in OA at the transcriptome level, including alternative splicing and potential regulatory mechanisms, and gene expression and molecular pathways, by analyzing RNA-seq data of knee cartilage tissues from OA patients with higher or lower OA score.
Method
Data Obtaining and Processing
We obtained raw fastq RNA-seq data of OA and NC articular cartilage from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) under accession no. GSE114007. Raw data from two kind of sequence platforms Illumina HiSeq 2000 and NextSeq 500 were first analyzed and compared, and then data produced from the NextSeq 500 platform was used for further study (Supplementary Table S1). In summary, fastq data were quality controlled using fastp (0.20.1), followed by mapping to genome sequence (GRCh37) using STAR (2.7.8a), then the mapped data were first processed consequently by featureCounts (2.0.3) and edgeR (3.36) to quantifying gene expression and identifying differential expressed genes, and second processed by rMATS (4.1.2) to quantifying alternative splicing and identifying differential alternative splicing events.
Differential Gene Expression Analysis
Followed by featureCounts processing, edgeR was used to identify DEGs using raw counts data. Genes with low expression were filtered using filterByExpr function in edgeR and most other steps were followed by the instructions.
Pathway Enrichment and GSEA Analysis
DEGs and genes related to differential alternative splicing events (DASEs) were used to perform function enrichment analysis by using the Metascape (www.metascape.org) website tool, and top 20 pathways with most significant P value were remained to visualization. GSEA analysis was performed by using R package fgsea (1.19.2) based on the order of all genes ranked by difference between OA and NC tissues. Pathways with false discovery rate (FDR) less than 0.05 were think as significant enriched pathways.
Alternative Splicing Analysis
There was a total of 103,231 ASEs detected by rMATS in these OA and NC articular cartilage tissues. Then we defined that PSI only calculated for ASEs with junction related reads more than 10 and removed ASEs with PSI values in less 75% samples, remaining 22,467 ASEs. For the annotated novel ASEs, we further removed ASEs with minimum junction counts less than 3 in all samples and finally included 15,305 ASEs in this study, including 12,092 known and 3,213 novel ASEs. Among these included ASEs, we identified 442 DASEs by using the delta percent splice in (PSI) cutoff more than 0.1 and FDR less than 0.05.
Annotation of Alternative Splicing Events
After downloading the GRCh37/GENCODEv19 version of the genome annotation file, the exons of each annotated transcript were extracted and sorted from 5’ to 3’ by genomic position. The splice junctions (SJs) in each splicing event were then identified based on their own location in the genome. These splicing event-related SJs were finally mapped to structured annotated transcripts to identify alternative splicing-related transcripts.
Statistics and Visualization
For all statistics and visualization tools were based on R software, among which packages gggplot2, VennDiagram, ComplexHeatmamap, and pheatmap were used.
Results
Data Analysis and Gene Expression Profiles in Osteoarthritis
By using two sequencing platforms (Illumina HiSeq 2000 and NextSeq 500), Fisch et al. 21 performed RNA-seq of 38 knee cartilage samples, 20 from OA patients (OA score 4) and 18 from normal control tissues (OA score 1, defined as NC), and stored this transcriptome data in the GEO public datasets (GSE114007). Through the analysis of this datasets, a lot of obvious differences between different sequencing platforms were found. Considering the intra-group differences of sequencing data using the NextSeq 500 platform are smaller, we only select these data for downstream analysis. The entire data analysis process includes data download, quality control, genome alignment, gene expression quantification and differential analysis, alternative splicing quantification and differential analysis, gene function enrichment and GSEA analysis, and enrichment analysis of RNA binding motifs ( Fig. 1 ).
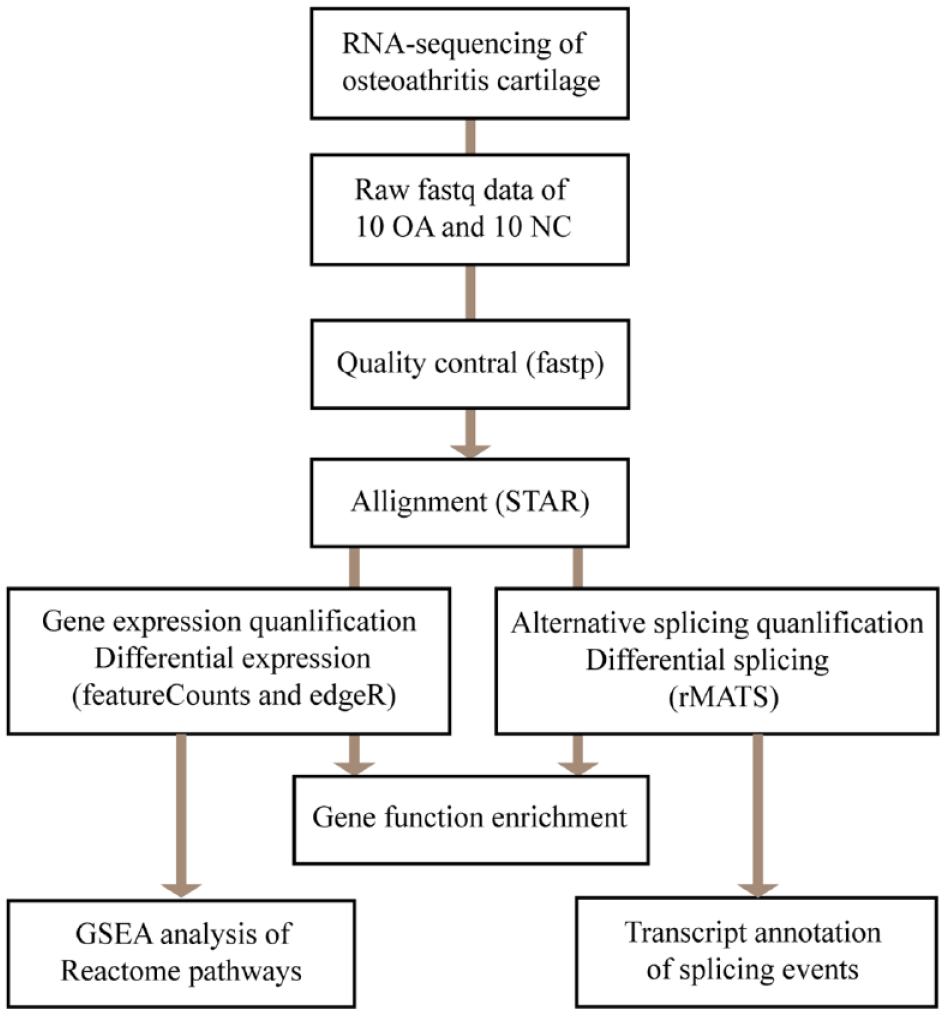
Flowchart of the study design. Raw RNA-seq data was obtained from the GEO database (GSE114007). After quality control, raw data were mapped to genome reference and used to generate bam file for each sample. Bam files were then used to perform expression and splicing quantification. Differential expressed genes and splicing events were then used to perform function enrichment analysis. OA = osteoarthritis; NC = normal control; GSEA = gene set enrichment analysis.
We used the FeatureCounts software for gene expression quantification and the edgeR for differential gene expression analysis, respectively. The results showed that a total of 17,935 genes were relatively high expressed in knee cartilage tissues. According to the screening cutoff|logFC| > 2 and FDR <0.05, differential expression analysis identified a total of 457 differential expressed genes (DEGs), including 266 down-regulated and 191 up-regulated DEGs ( Fig. 2A , Supplementary Table S2). Cluster analysis was performed for the 20 included samples based on these DEGs, and results showed that the OA or NC samples were clustered together well, indicating that the differential expression analysis was reliable ( Fig. 2B ). The up-regulated genes such as TNFSF15 and ST6GALNAC5 and the down-regulated genes such as ADM and JUN in Fisch’s analysis were also identified with the same changes in our analysis, which in turn also demonstrated that our analysis was reliable. In addition, among the most significant differential expressed genes (ranked by FDR), we also found that the expression of genes such as TGFBI, ASPM, and TYMP were upregulated, and the expression of genes such as IRE2, PIGA, and MAFF were significantly down-regulated in OA ( Fig. 2C ).
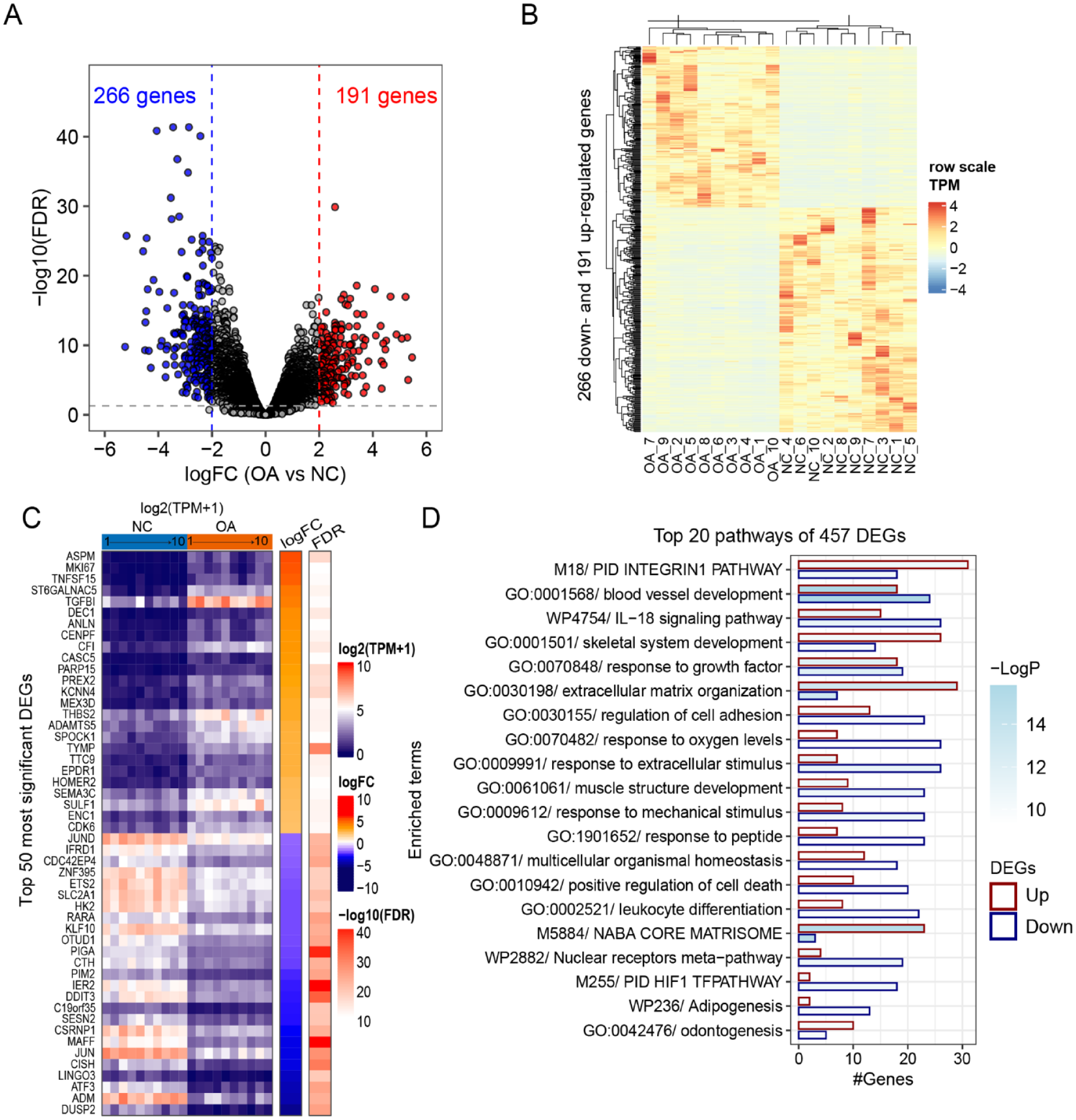
Profiles and variants of gene expression in osteoarthritis. (
By using website tool Metascape to perform function enrichment analysis for all DEGs, we found that pathways mainly enriched by up-regulated DEGs were “GO:0030198 extracellular matrix organization,” “M5884 NABA CORE MATRISOME,” “M18 PID INTEGRIN1 PATHWAY,” “GO:0001501 skeletal system development,” and “GO:0042476 odontogenesis”, while pathways mainly enriched by down-regulated DEGs were “GO:0009612 response to mechanical stimulus,” “GO:0009991 response to extracellular stimulus,” “GO:0070482 response to oxygen levels,” “GO:1901652 response to peptide,” “GO:0061061 muscle structure development,” “M255 PID HIF1 TFPATHWAY,” “WP2882 Nuclear receptors meta-pathway,” and “WP236 Adipogenesis” ( Fig. 2D ).
Not surprisingly, the expression of most genes in the extracellular matrix (ECM) related pathways was significantly upregulated in OA, suggesting a severe imbalance between ECM synthesis and degradation. Nonetheless, in the “Extracellular matrix organization” or “CORE MATRISOME” pathway, in addition to the elevated expression of most genes, the expression of genes such as SOX9, ELF3, TNFRSF1B, ICAM1, FGL2, and SPOCK3 were significantly decreased (Suppl. Fig. S1). For example, SOX9 transcriptionally activates the genes for cartilage-specific structural components and secures chondrocyte lineage commitment by promoting cell survival, which is required for chondrogenesis 25 and articular cartilage healthy. 26 In patients with osteoarthritis, however, SOX9 is reduced in the articular cartilage, which makes it a potential target for OA treatment.27,28 Moreover, the reduction in the expression of genes in those “response to” pathways indicates that the stress-response ability of chondrocytes is weakened and the degradation will continue to accumulate, which may be one of the reasons why the OA disease process is difficult to block and develop continuously.
Gene Set Enrichment Analysis and Pathways Changed in Osteoarthritis
Pathway enrichment analysis for significant DEGs can reliably study the pathway changes in OA; however, many pieces of useful information may be missed inevitably. Therefore, we performed GSEA by making full use of differential expression result of all genes detected, for the 1,068 molecular pathways in the “Reactome” project. 29 The results showed that compared with NC tissues, OA tissues had 29 significantly up-regulated (NES > 0 and adjust P-value < 0.05) pathways, and 75 significantly downregulated (NES<0 and adjust P-value <0.05) pathways ( Fig. 3A ). Among the most significant 30 pathways (15 upregulated and 15 downregulated), we found that up-regulated pathways in OA mainly included “Extracellular matrix (ECM),” “Collagen” related pathways ( Fig. 3B ). Interestingly, pathways related to ECM or collagen formation and degradation were simultaneously enriched in OA ( Fig. 3C ), indicating that ECM or collagen-related synthesis in OA is a dynamic process, rather than a complete degeneration process as conventionally thought.7,30 During the dynamic alteration of cartilage in the osteoarthritis process, the imbalanced ECM loses its normal structure and composition and the cartilage loses its integrity. 31 In cartilage, chondrocytes are responsible for the biogenesis and maintenance of the ECM, including proteins, glycoproteins, and proteoglycans. By using scRNA-seq, Ji et al. 22 found that chondrocytes are different in variable stage OA tissues, and can be classified into 7 subgroups with distinguished functions in the pathological cartilage, including proliferative chondrocytes (ProCs), prehypertrophic chondrocytes (preHTCs) and hypertrophic chondrocytes (HTCs), fibrocartilage chondrocytes (FCs) and three novel populations named effector chondrocytes (ECs), regulatory chondrocytes (RegCs), and homeostatic chondrocytes (HomCs). HTCs and FCs are characteristic with unfavorable genes and associated with progression of OA process, while FCs also expressed genes enriched for vasculature development and extracellular matrix (ECM) organization and enriched in late-stage OA. 22 Thereby, conditions and changes in pathways are responsible for differentiation or switch to advanced subtypes of chondrocytes, which could play a role in the initiation or progression of OA. For example, we found that NCAM-, PDGF-, and IGF-related signal transduction pathways and protein phosphorylation pathway were upregulated in OA tissues ( Fig. 3B ). In the process of chondrogenic differentiation, NCAM is expressed in prechondrogenic cells and increased during cell condensation. 32 Altered IGF-1 levels was reported to affect the development of OA but also be potentially the result of the pathophysiological OA process.33,34
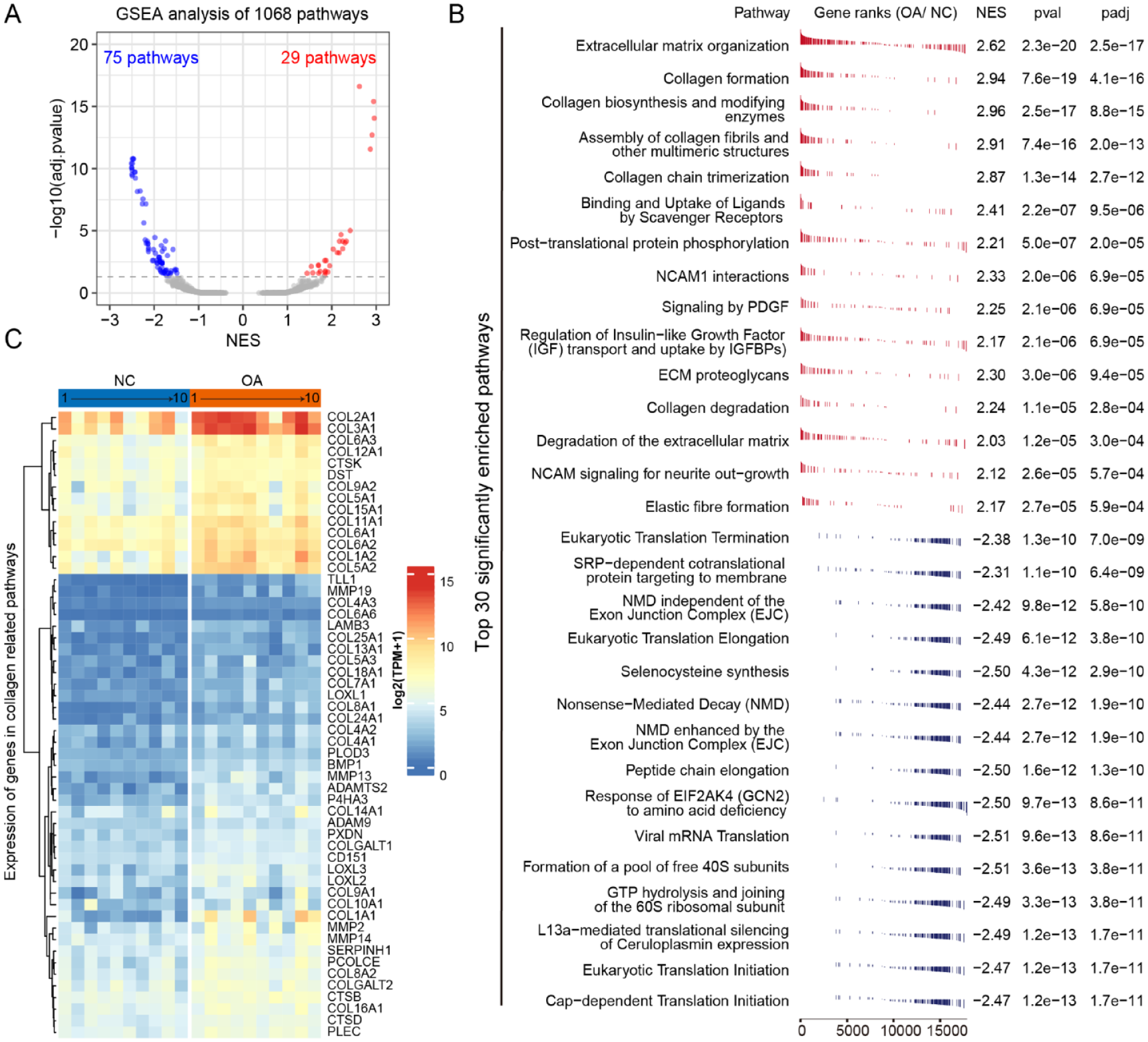
Gene set enrichment analysis identify pathways changed in osteoarthritis. (
The downregulated pathways in OA mainly include protein translation-related pathways ( Fig. 3B ). All significantly differential enriched pathways were further classified and summarized (Supplementary Table S3). It was found that downregulated pathways also included transcription, immunity, PI3K/AKT, circadian, and other related pathways. Downregulated expression of genes in these translation-related pathways means lower translation efficiency and quality control system. Translation errors of proteins in chondrocytes causing the accumulation of error proteins in the endoplasmic reticulum (ER) can consequently trigger unfolded protein response (UPR)-initiated cell death, which may be a condition to promote chondrocytes death in OA. 35 Moreover, repression of transcription was enriched in FOXO, PERK, NGF, and TFAP2 family regulated genes (Supplementary Table S3). Intriguingly, FOXO-mediated genes related to cell death were also repressed in OA, which means transcript factors regulated gene expression also contribute to OA progression.
Alternative Splicing in Osteoarthritis
Alternative splicing analysis was performed by using the rMATS software and five splice types including alternative 3’ splice site (A3SS), alternative 5’ splice stite (A5SS), skipped exon (SE), mutually exclusive exons (ME), and retained intron (RI) were analyzed ( Fig. 4A ). A total of 103,231 alternative splicing events detected in cartilage tissues and 15,305 ASEs within 3,048 genes remained after filtering by percentage samples with value more than 75 percentage and junction reads more than 3 for novel ASEs. The number of ASEs in different splice types were 1,063, 832, 1,972, 1,095, and 10,343 for A3SS, A5SS, MXE, RI, and SE, respectively, among of which 12,092 ASEs were genome annotated known and 2,313 were novel. According to the screening cutoff delta PSI > 0.1 and FDR < 0.05, we identified 442 differential alternative splicing events (DASEs) in OA, including 196 DASEs with PSI up-regulated and 246 down-regulated ( Fig. 4B and C , Supplementary Table S4). There were 70 most significant DASEs within 50 genes identified with delta PSI > 0.2 and FDR < 0.0001 in OA ( Fig. 4D ), including 11 known OA-associated genes such as IL16, CIRBP, PRG4, PDK1, ITGA5, LRP1, ABI3BP, PDK1, SHMT2, and THBS3 ( Fig. 4D and E ). For example, IL16 (interleukin-16) have been associated with various disease states, and its activity is dysregulated in synovial fibroblasts of individuals with rheumatoid arthritis. 36 Recently, the association between genetic polymorphisms in the gene encoding IL-16 and susceptibility to primary knee osteoarthritis have been identified in the Chinese Han population. 37 Thereby, the splicing of IL16 exon 3 might also be associated with OA process.
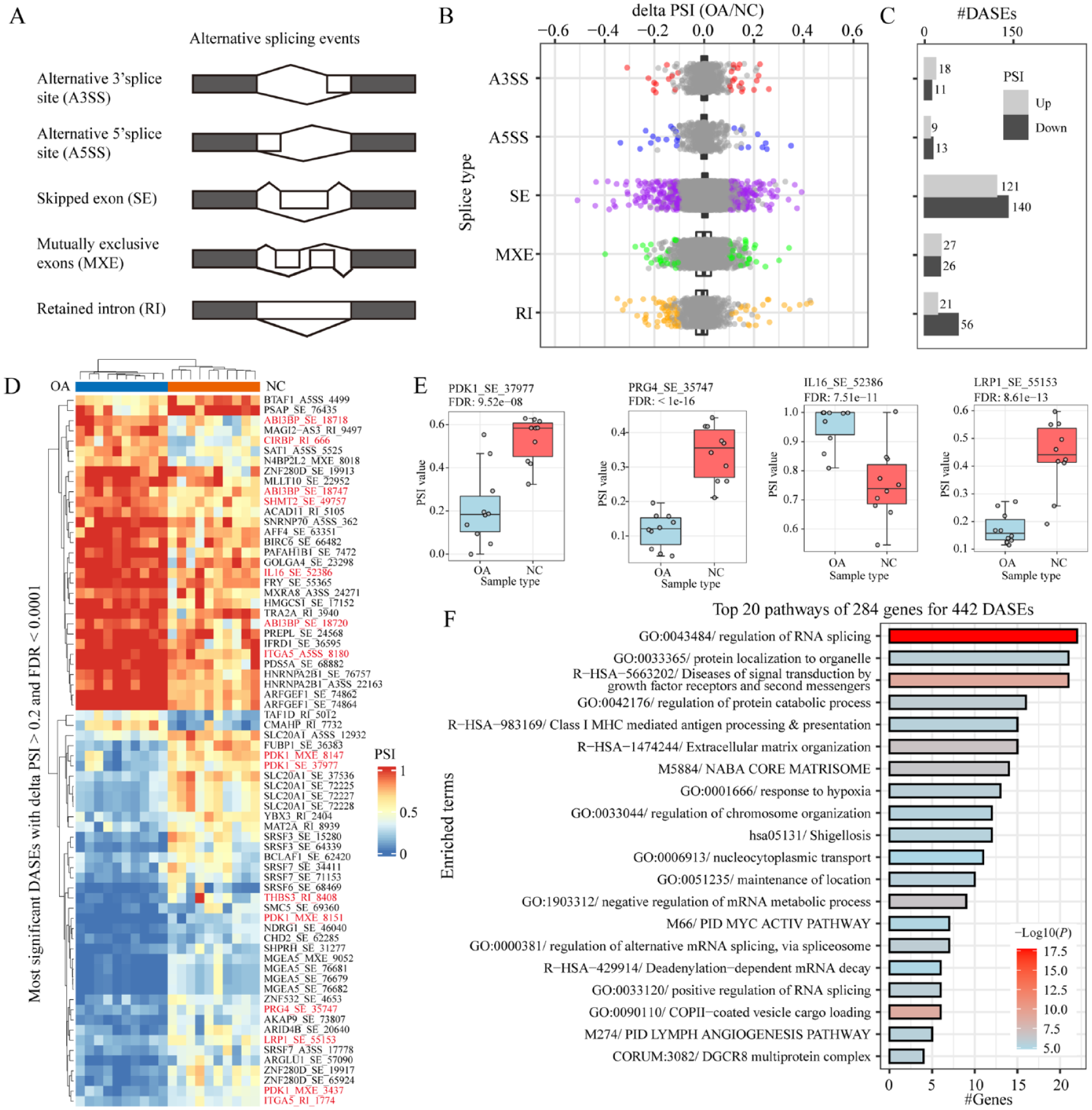
Alternative splicing in osteoarthritis. (
Furthermore, enrichment analysis for these DASEs was performed by using the Metascape tool. We found that “RNA splicing” was the most significant enrichment pathway for those DASEs, followed by pathways like “Protein localization and catabolic process,” “Diseases of signal transduction by growth factor receptors,” “Extracellular matrix organization,” “Core matrisome,” and “Response to hypoxia” ( Fig. 4F and Suppl. Fig. S2). These results indicated that alternative splicing of genes in these pathways and consequent perturbation of pathway functions might contribute to OA occurrence and progression. For example, fibroblast growth factor receptors (FGFR1-4) and consequent signaling is a prerequisite for the correct development and homeostasis of articular cartilage and aberrant FGF signaling contributes to the onset and progression of osteoarthritis. 38 As a member of the pathway “Diseases of signal transduction by growth factor receptors,” alternative splicing of FGFR1 might also involve in OA. Intriguingly, the result in our study that splicing factors are susceptibility to splicing was also found in several other studies,39,40 indicating that splicing of splice factors response to stress conditions may also function in OA process. In summary, these results showed that alternative splicing events changed in OA, which might also draw contribution to the onset and progression of OA.
Effect of Alternative Splicing in Osteoarthritis
Alternative splicing can draw an effect on gene expression and function by regulating the stability and coding ability of splicing transcripts or by changing the coding sequence of splicing transcripts and protein sequences. We found that differential alternative splicing events in OA were also enriched in ECM or collagen-related pathways ( Fig. 4F ), which means that alternative splicing play a role in chondrocyte in response to OA conditions such as excessive mechanical loading. We first analyzed the effect of AS to gene expression. By using Venn plot, however, only 27 genes were found in both 284 protein coding genes of 442 DASEs (delta PSI > 0.1 and FDR < 0.05) and 1,669 moderate DEGs (logFC > 1 and FDR <0.05) in OA, including 7 upregulated and 20 down-regulated DEGs ( Fig. 5A ). Among these 27 genes, only FN1, COL6A3, COL11A1, and COL1A2 were ECM or collagen related (Supplementary Fig. S3), indicated that alternative splicing and gene expression changes may play synergistic effect in the ECM regulation. Genewise Pearson correlation analyses were performed between PSI value of ASEs and gene expression for these 27 genes ( Fig. 5B ), we found that 16 genes showed significant correlations between AS and gene expression (P < 0.01). Furthermore, there were 78 genes were found in both 284 DASE-related genes and 4,197 mild DEGs (logFC >0.5 and FDR <0.05) in OA, including 15 –C upregulated and 63 downregulated DEGs ( Fig. 5C ). These data indicated that alternative splicing can draw a regulation effect, especially negative effect, on gene expression, but only for a small number of DASEs.
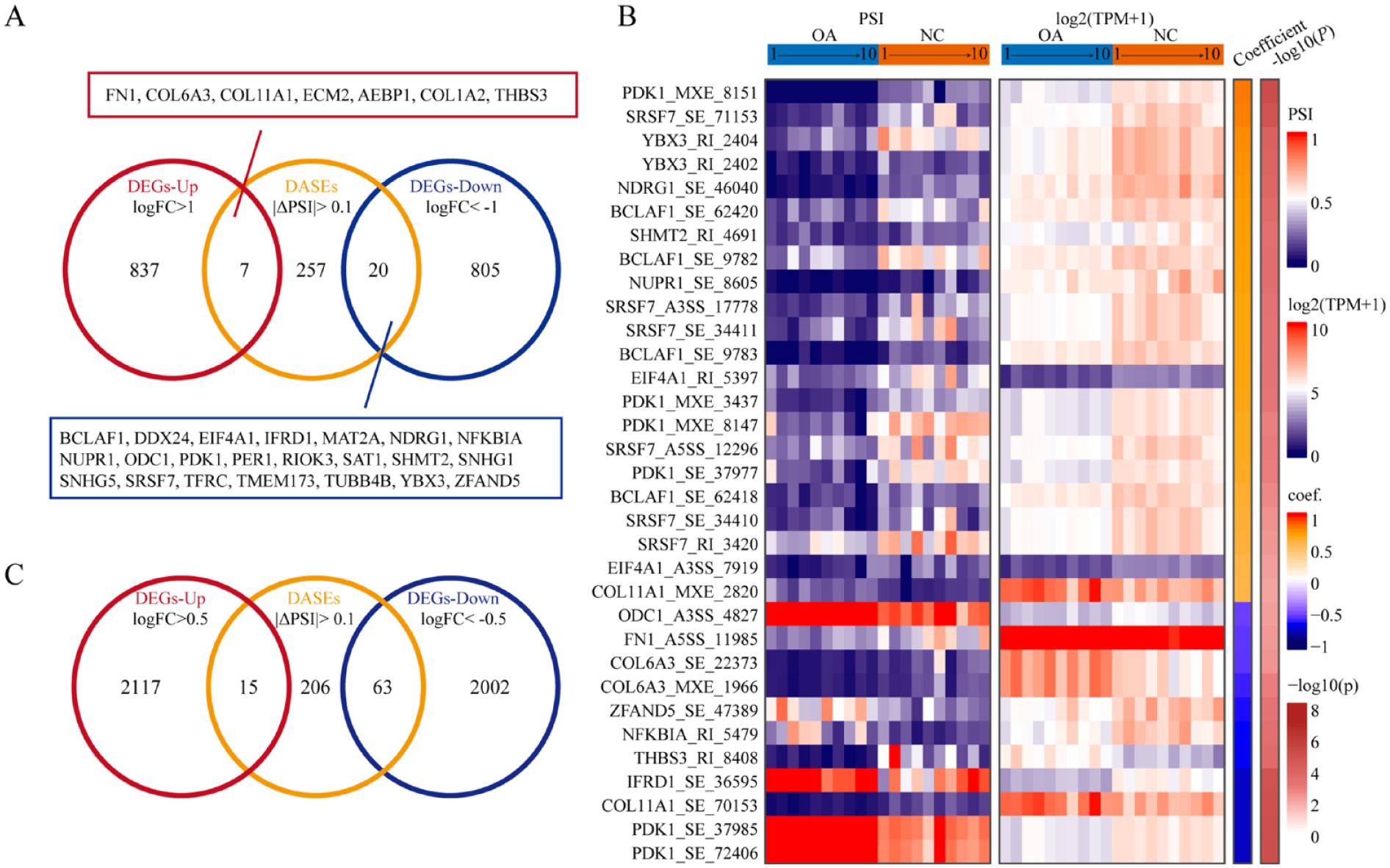
Effects of alternative splicing in osteoarthritis. (
To address gene regulation by alternative splicing, we next checked that if DASEs regulate coding sequences of splicing transcripts or protein products by mapping the splicing exons to transcripts in different biotype. Splicing exons were mapped to transcripts by genome locations. We classified biotype of transcripts into two groups named protein coding (PC) and non-protein coding (NPC, including nonsense-mediated decay, processed transcripts, retained intron, and antisense). Each ASE (alternative splicing events) can be allocated with two types of transcripts named Splicing exon In (SI) and Splicing exon Out (SO) transcripts. The results showed that there were 192 DASEs annotated with transcripts in both SI and SO transcripts, including 43 DASEs annotated with only PC transcripts, 74 DASEs annotated with NPC transcripts in both SI and SO transcripts, 55 DASEs annotated with NPC transcripts in SI transcripts but not in SO transcripts, and 20 DASEs annotated with NPC transcripts in SO transcripts but not in SI transcripts. Moreover, there were 250 DASEs without annotated transcript in SI or SO transcripts, including 142 DASEs annotated with no SI transcript, 64 DASEs annotated with no SO transcript, and 44 DASEs annotated with nether SI nor SO transcript (Supplementary Table S4).
Among the 55 DASEs annotated with NPC transcripts in SI transcripts and with PC transcripts in SO transcripts, there were 14 DASEs characterized with up-regulated PSI in OA, including splicing factors (SFs) PNISR, TRA2A, RBMX, SON; transcription factors (TFs) SOX5; and ECM related gene MXRA8, which means protein production of these genes reduced due to alternative splicing in OA. Inversely, there were 41 DASEs characterized with down-regulated PSI in OA, including SFs such as SRSF3, SRSF6, SRSF7, U2AF1; TFs like BCLAF1, BTAF1, MLF2, PTMA, NUPR1, DMTF1, and AEBP1; translation factors like EIF4A1, EIF5; and ECMs like FN1, ITGA5, OGT, TNS1, PLOD2, and ITPR2, which means protein production of these genes expanded due to alternative splicing in OA (Supplementary Table S4).
Furthermore, there were 43 DASEs annotated with only PC transcripts in both SI and SO transcripts, especially for genes function in ECM, including COL11A1, COL16A1, COL6A3, CAST, DCN, P4HA2, PDK1, and PRG4. For example, MXE splicing of COL11A1 exon 6 and exon 7 results in two splicing isoforms COL11A1-001 (with exon 6) and COL11A1-202 (with exon 7). Upregulated PSI of MXE splicing of COL11A1 means increasing expression of COL11A1-202 in OA ( Fig. 5D ). ES splicing of COL16A1 exon 43 results in COL16A1-005 which is shorter than COL16A1-001. Upregulated PSI of COL11A1 splicing means increasing expression of COL11A1-001 in OA. These results together indicated that splicing of ECM related genes and consequently isoforms switching might play a role in OA.
Regulation of Alternative Splicing in Osteoarthritis
Alternative splicing can be modulated by many factors, including expression and modification of RNA binding proteins (RBPs), transcription elongation and RNA structure. Here, we will focus on studying the regulation effect of RBPs to alternative splicing. In all, 521 RBPs were collected from pathways or gene sets annotated with “splicing,” “splice,” “mRNA binding,” or “spliceosome” in the MSigDB (https://www.gsea-msigdb.org/). Among these RBPs, we identified 22 differential expressed RBPs in OA (logFC > 1 and FDR < 0.05), including 6 up-regulated RBPs such as TIA1, NOVA1 and 16 downregulated RBPs such as SRSF7 and RBM47 ( Fig. 6A ). We further examined if DASEs in OA enriched with motifs of any RBPs by using rMAPS (http://rmaps.cecsresearch.org/). ASEs without any splicing change in OA (FDR > 50%, maximum PSI > 15% and minimum PSI < 85%) are compiled and used as control (non-regulated ASEs). Results showed that DASEs with exon inclusion in OA were mainly enriched to motifs of SRSF1, PCBP1, PCBP3, PTB, LIN28A, and ZCRB1, while DASEs with exon skipping in OA were mainly enriched to motifs of HuR, SRSF10, CPEB4, PABPC1, RALY, ZC3H14, and HNRNPC. Moreover, motifs of RBMS3, SRp20, and TIA1 were enriched for both DASEs with exon skipping and inclusion ( Fig. 6B and Suppl. Fig. S4). Integration of these data, we found that only TIA1 was deferentially expressed and significantly enriched by DASEs in OA, which means TIA1 regulated DASEs might contribute to the OA process.
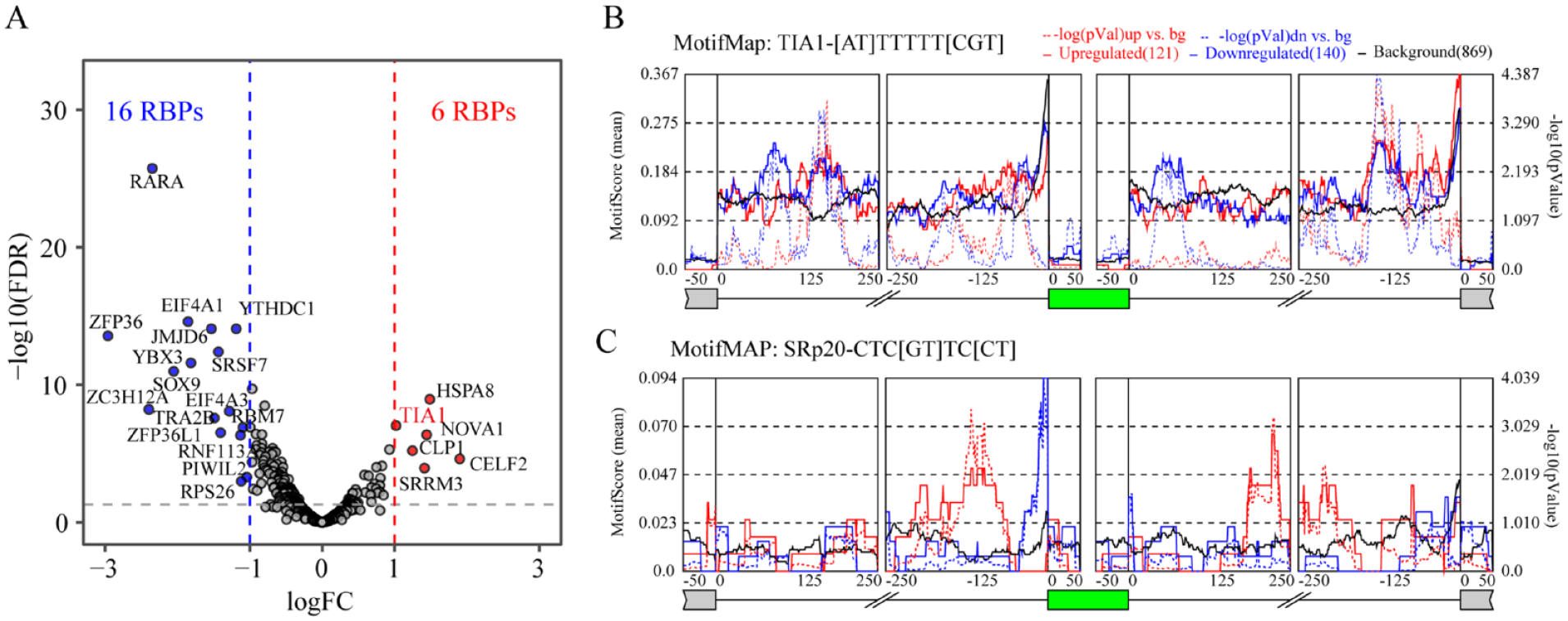
Potential RNA binding proteins in the regulation of alternative splicing in osteoarthritis. (
Discussion
In this study, we have performed an integrative analysis of RNA-seq data to identify transcription changes in OA, including differential gene expression, pathways and alternative splicing. Results showed that DEGs in OA mainly enriched to ECM and collagen pathways. GSEA analysis identified additional pathways such as translation, transcription, immunity, PI3K/AKT, and circadian in OA. Most importantly, we have identified hundreds of DASEs in OA, including previously reported and many others newly detected. Function enrichment of these DASEs showed that they also involved in OA process. Among this DASEs, gene function can be affected by several ways, including regulation of gene expression and alternative usage of coding sequence. Finally, by integrating expression data of RBPs and motif enrichment of DASEs, we found that TIA1 may be one of significant factors affecting alternative splicing in OA.
Biological pathways within a joint are mechanosensitive and the amelioration of biological mechanical stress can alleviate OA progression, which make it a potential means of intervention. Exposure of articular cartilage to excessive mechanical loading is deeply involved in the pathogenesis of osteoarthritis through gremlin-1, which antagonizes induction of anabolic genes such as Sox9, Col2a1, and Acan by bone morphogenetic proteins. 41 Other cellular stresses, such as hypoxia, nutrient deprivation or oxidative stress in articular cartilage, can also affect the synthesis and secretion of ECM proteins and induce cell death, which is associated with the pathogenesis of osteoarthritis. 35 The results of bioinformatics analysis based on microarray data showed that differentially expressed genes in OA were significantly enriched in “response to” related pathways. 42 In our results, we also found that some downregulated genes in OA were also enriched in these pathways ( Fig. 2D ). The decreased expression of genes in these response pathways indicated more likely that the chondrocytes are susceptive to stimulation or transcription factors are reduced in response to stimulation, but less likely that stimulation are less in OA than normal controls. In addition to that, GSEA results also showed that downregulated genes were enriched to translation, transcription and immunity related pathways. Intriguingly, these changes in pathways indicated suppression status of cell activity, which often appeared when integrated stress response (ISR) pathway was activated. The ISR system is a conserved intracellular signaling network that helps the cell and organism to adapt to different environmental and pathological conditions, including protein homeostasis defects, nutrient deprivation, and oxidative stress. 43 The ISR can restores balance by reprogramming gene expression and maintain cell health. In OA, chondrocytes often suffering stress conditions such as mechanic overloading, nutrients deprivation, and hypoxia stress. Therefore, dysfunction of ISR system in OA may be a reason of chondrocytes death and disease progression and blocking or ameliorating the related stress conditions may slow down the continuous progression of OA.
Moreover, GSEA results also showed that circadian pathway was downregulated in OA. Dysfunction of brain-controlled rhythmic system may affect risk factors for OA in several ways, including the link between the circadian clock and metabolism. 44 For example, abnormalities in circadian rhythms can affect OA by increasing obesity risk or affecting the balance of ECM synthesis and degradation.45,46 Dysregulation of circadian rhythm may also affect the physical activity cycles, resulting in inappropriate responses of local joint tissues to rhythmic challenges, such as mechanical loads associated with daily movement. 44 This rhythm-related functional dissociation not only leads to increased susceptibility of knee cartilage to injury, but may also affect the repair process of injury, which may be important factors in the continued progression of OA.
Alternative splicing often involved in regulation process of mRNA expression and gene function, which may involve in OA through several aspects. For example, Superficial Zone Protein (SZP) is expressed by the superficial zone chondrocytes and involved in boundary lubrication of the articular cartilage surface. Alternative splicing of SZP and splicing out of the heparin-binding domain regulated by TGF-β1 may regulate SZP interaction with heparin sulfate and components in the ECM of articular cartilage. 47 The major proteoglycan of articular cartilage aggrecan is a substrate for ADAMTS4. Alternative splicing and using of 3’ splice site within the 9 exon of ADAMTS4 resulting a smaller transcript missing 161 bp from the 5’ end of exon 9 and protein with new C-terminal domain, which may alter its substrate specificity. 48 Despite this, only a small number of cases about alternative splicing and its effects in OA were studied. Recently, an integrative transcriptome analysis of long non-coding RNA and alternative splicing in OA were performed and several differential alternative splicing events were identified, 49 including ASEs validated in our study. Our data further showed that alternative splicing in OA often occurred in genes related to ECM and collagen pathways as well as splicing factors. In most conditions, alternative splicing of these genes can cause disruption of transcripts, while in some cases AS may cause AS-associated NMD and some others may cause alternative usage of coding sequences. For example, MXRA8 may play a function in inhibiting osteoclastogenesis and promote proliferation and maturation of growth plate chondrocytes and plays a role in cartilage formation. Thereby, alternative splicing of MXRA8 and consequently reduced protein production may be a reason of cartilage lesions and bone remodeling. ITPR2 encodes inositol 1,4,5-triphosphate receptor (IP3R) type 2, belonging to the IP3R family, which function as intracellular Ca+ channels residing in the membrane of endoplasmic reticulum (ER). ITPR2 was reported function in the regulation of apoptosis; 50 therefore, alternative splicing of ITPR2 and consequently expanded protein production might contribute to excessive chondrocyte apoptosis and cartilage lesions in patients with OA. 51 Our results expanded the alternative splicing profiles and their effects in OA.
To further understand the occurrence and development of OA, our results provide favorable information to support the development of more effective interventions in the future. Compare to previous study in gene expression and pathways or alternative splicing, our data provide some beneficial results such as perturbed genes in stress and response pathways, the profiles, effects and mechanisms of alternative splicing in OA.21,49 However, this study also has several limitations. First, our analysis only included two conditions of OA process, including OA score 1 and score 4, which might be in-sufficient to identify OA progression associated changes. Furthermore, our analysis only focused on molecular changes in transcription level, which cannot illustrate completely alternative splicing changes in OA and multi-omics might be needed in the future study.
Supplemental Material
sj-docx-1-car-10.1177_19476035231154511 – Supplemental material for Integrated Analysis of Transcriptome Changes in Osteoarthritis: Gene Expression, Pathways and Alternative Splicing
Supplemental material, sj-docx-1-car-10.1177_19476035231154511 for Integrated Analysis of Transcriptome Changes in Osteoarthritis: Gene Expression, Pathways and Alternative Splicing by Congming Li, Pengli Wei, Lei Wang, Qiang Wang, Hong Wang and Yangjun Zhang in CARTILAGE
Supplemental Material
sj-xlsx-2-car-10.1177_19476035231154511 – Supplemental material for Integrated Analysis of Transcriptome Changes in Osteoarthritis: Gene Expression, Pathways and Alternative Splicing
Supplemental material, sj-xlsx-2-car-10.1177_19476035231154511 for Integrated Analysis of Transcriptome Changes in Osteoarthritis: Gene Expression, Pathways and Alternative Splicing by Congming Li, Pengli Wei, Lei Wang, Qiang Wang, Hong Wang and Yangjun Zhang in CARTILAGE
Footnotes
Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Study conception and design: Congming Li, Hong Wang, and Qiang Wang. Analysis and interpretation of data: Congming Li, Yangjun Zhang, Pengli Wei and Lei Wang. Drafting of the article: Congming Li Yangjun Zhang.
Acknowledgments and Funding
We appreciate the technical support from Xudong Zhang. The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.