
Abstract
Objective
Osteoarthritis (OA) is a severe and common degenerative disease; however, the exact pathology of OA is undefined. Our study is designed to investigate the underlying molecular mechanism of OA with bioinformatic tools.
Design
Three updated GEO datasets: GSE55235, GSE55457, and GSE82107 were selected for data analyzing. R software was utilized to screen and confirm the candidate differentially expressed genes in the development of OA. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway were performed to identify the enriched GO terms and signaling pathways. Protein and protein interaction (PPI) models were built to observe the connected relationship among each potential protein.
Results
A total of 113 upregulated genes and 161 downregulated genes were found by integrating 3 datasets. GO enrichment indicated that cell differentiation, cellular response to starvation, and negative regulation of phosphorylation were important biological processes. KEGG enrichment indicated that FoxO, IL-17 signaling pathways, and osteoclast differentiation mainly participated in the progression of OA. Combining the molecular function and PPI results, ubiquitylation was identified as a pivotal bioactive reaction involved in OA.
Conclusion
Our study provided updated candidate genes and pathways of OA, which may benefit further research and treatment for OA.
Keywords
Introduction
Osteoarthritis (OA) is deemed a common disease that brings much health, social, and economic burden to the world. 1 The global morbidity of OA has reached a high level in recent years. In people over the age of 60 years, 10% of males and 19% of females suffer from OA. 2 Focusing on different body parts, the incidence of knee OA peaks at 0.24% of the world population, followed by hip OA and hand OA. 3 During the development of OA, erosion of cartilage, sclerosis of subchondral bone, and synovial inflammation comprise the most key pathogenic links that result in pain, stiffness, and deformity of joints. 4
The current treatment strategy for OA is a stepwise scheme, in which different procedures are applied at different stages. 5 Although present therapies have achieved some satisfactory results, numerous patients cannot acquire complete relief of various symptoms. With the advantages of bioinformatic technology, research for genes and proteins has emerged as potential treatment targets. 6 As a representative tool, National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database stores enormous data about gene expression datasets, original series, and platform records. By analyzing and comparing gene expression level from each microarray dataset, different expression genes (DEGs) can be filtered and studied. Subsequently, alterations at the microcosmic aspect such as genetic or epigenetic may offer more information about the disease. Previous studies have uncovered lots of biomarkers that benefit the diagnosis, treatment, and prognosis of OA. However, with the rapid speed of updates in bioinformatics data, new analyzing results are required to explore the underlying mechanisms of OA.
In the present bioinformatic analysis, based on 3 recently updated datasets: GSE55235, 7 GSE55457, 7 and GSE82107, 8 the authors screened and obtained DEGs from knee synovium tissue between normal and OA patients. Performing Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis elucidated potential gene ontology terms and signaling pathways. Moreover, the interacted relationship among these identified proteins was examined with help of PPI network. Our study aimed to investigate the updated bioinformation during the pathogenesis of OA and provide newly candidate biomarkers and molecular mechanisms.
Materials and Methods
Data Downloading
NCBI GEO datasets were used to screen and download 3 identified GSE series: GSE55235, GSE55457, and GSE82107 (https://www.ncbi.nlm.nih.gov/gds/). All data were collected from human synovium tissue. GSE55235 and GSE55457 both contained 10 samples of normal (N) and 10 samples of OA, based on GPL96 [HG-U133A] Affymetrix Human Genome U133A Array platform. GSE82107 contained 7 samples of N and 10 samples of OA, based on GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array platform. Statistical analyses were performed using R software (https://www.r-project.org/).
Identification of DEGs
Linear Models for Microarray Data package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) was applied for screening the DEGs between N and OA samples and drawing the volcano map under R environment. Batch normalization was performed with Surrogate Variable Analysis package (http://www.bioconductor.org/packages/release/bioc/html/sva.html). Heatmap package (http://www.bioconductor.org/packages/release/bioc/html/heatmaps.html) was installed to produce the heatmap. The cutoff value of log2 fold change was 1 and adjusted P value was set as 0.05.
GO and KEGG Enrichment
Gene Ontology (GO) consists of 3 major components: biological processes (BP), cellular component (CC), and molecular function (MF), which annotates gene functions and mutual connections. Kyoto Encyclopedia of Genes and Genomes (KEGG) is a wide database resource that discloses high-level functions and utilities of the biological system. ClusterProfiler package: (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html) was used to analyze GO and KEGG enrichment. to DOSE, COLORSPACE, STRINGI, and PATHVIEW package was utilized to build a visualization of the results. P value less than 0.05 was considered as the cutoff.
PPI Network Integration
Protein and protein interaction (PPI) network provide detailed connection numbers and strength among each selected protein. Search Tool for the Retrieval of Interacting Protein (https://string-db.org/) was applied to potential PPI network. Confidence score was set as 0.7 and disconnected nodes in the network were hidden. Cytoscape (https://cytoscape.org/) was used to visualize the interaction between each protein. R software was conducted to calculate the nodes number and identify the key genes.
Results
Identification of DEGs
To screen the potential DEGs, microarray data from GSE55235, GSE55457, and GSE82107 were collected and analyzed. After filtration and normalization, a total of 274 DEGs that comprised 113 upregulated genes and 161 downregulated genes were identified. All DEGs between N and OA were showed in heatmap according to different source (
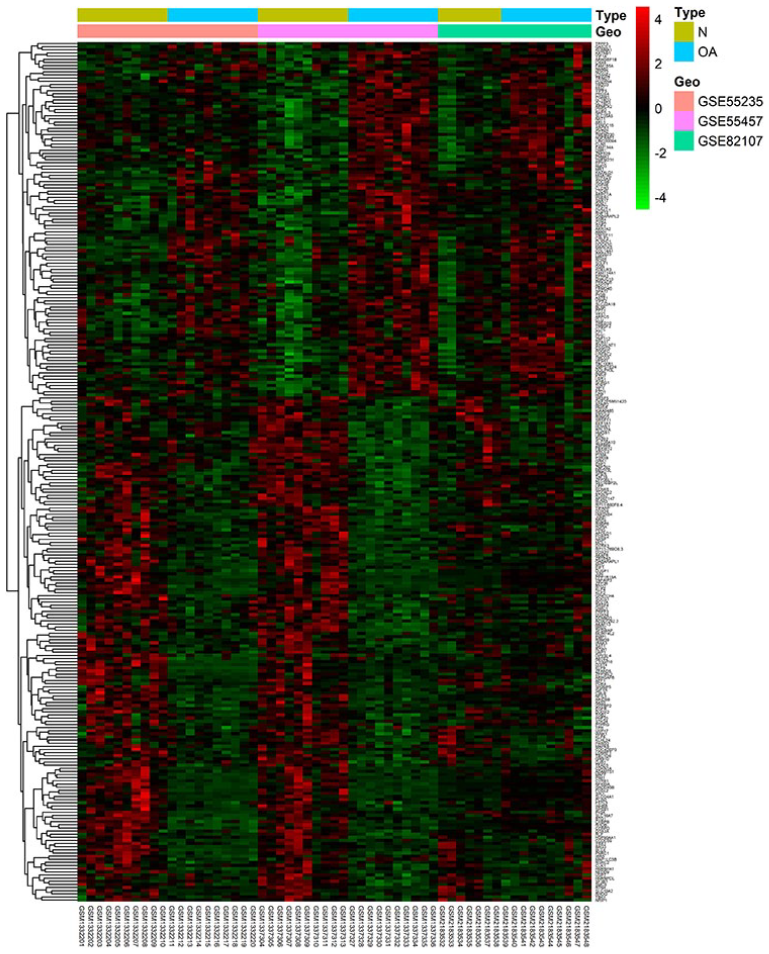
Heatmap of the differentially expressed genes between osteoarthritis (OA) and normal samples from 3 Gene Expression Omnibus (GEO) databases. The horizontal axis represents the sample numbers and the vertical axis represents the gene names. The red rectangles represent upregulated genes and green rectangles represents downregulated genes.
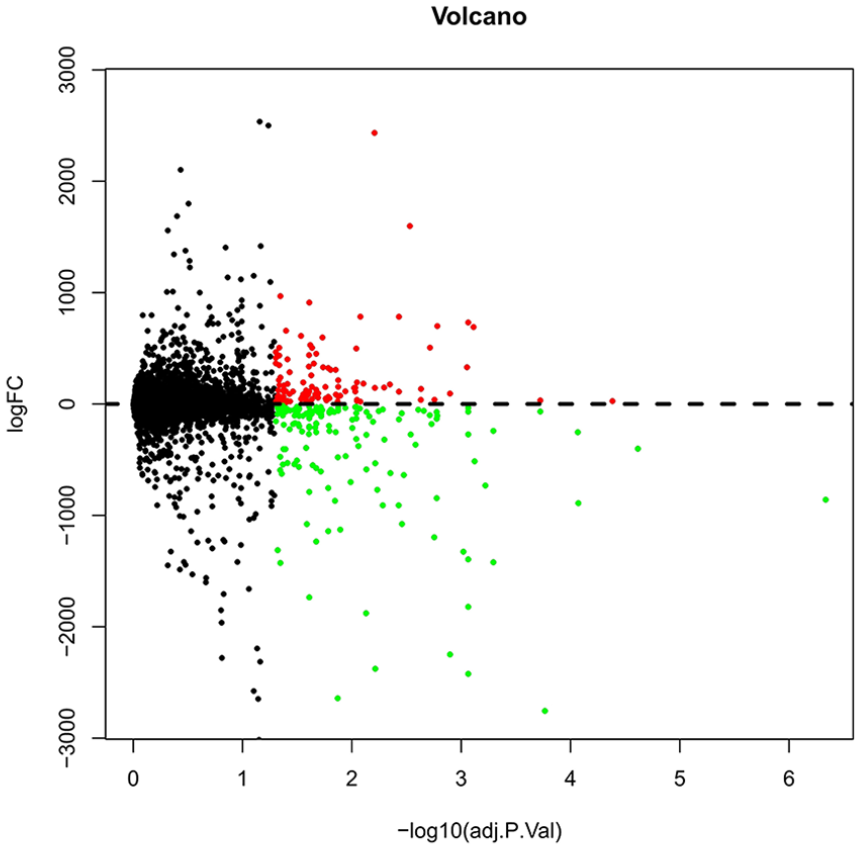
Volcano graph of the differentially expressed genes between osteoarthritis (OA) and normal samples. The red points represent the upregulated genes, green points represent downregulated genes, and the black points represent no significantly changed genes.
GO Functions
GO function enrichment analysis showed that DEGs between N and OA samples mainly participated in cell differentiation and starvation process. The top 10 enriched BP terms were fat cell differentiation (GO:0045444), cellular response to starvation (GO:0009267), response to starvation (GO:0042594), cellular response to external stimulus (GO:0071496), cellular response to nutrient levels (GO:0031669), negative regulation of phosphorylation (GO:0042326), negative regulation of protein phosphorylation (GO:0001933), cellular response to extracellular stimulus (GO:0031668), cellular response to peptide (GO:1901653), stress-activated protein kinase signaling cascade (GO:0031098). The enriched CC terms were ficolin-1-rich granule lumen (GO:1904813) and nuclear speck (GO:0016607). The enriched MF terms were ubiquitin-like protein ligase binding (GO:0044389) and ubiquitin protein ligase binding (GO:0031625). Top 10 enrichment on BP and all enrichment on CC and MF were shown in a bubble graph (
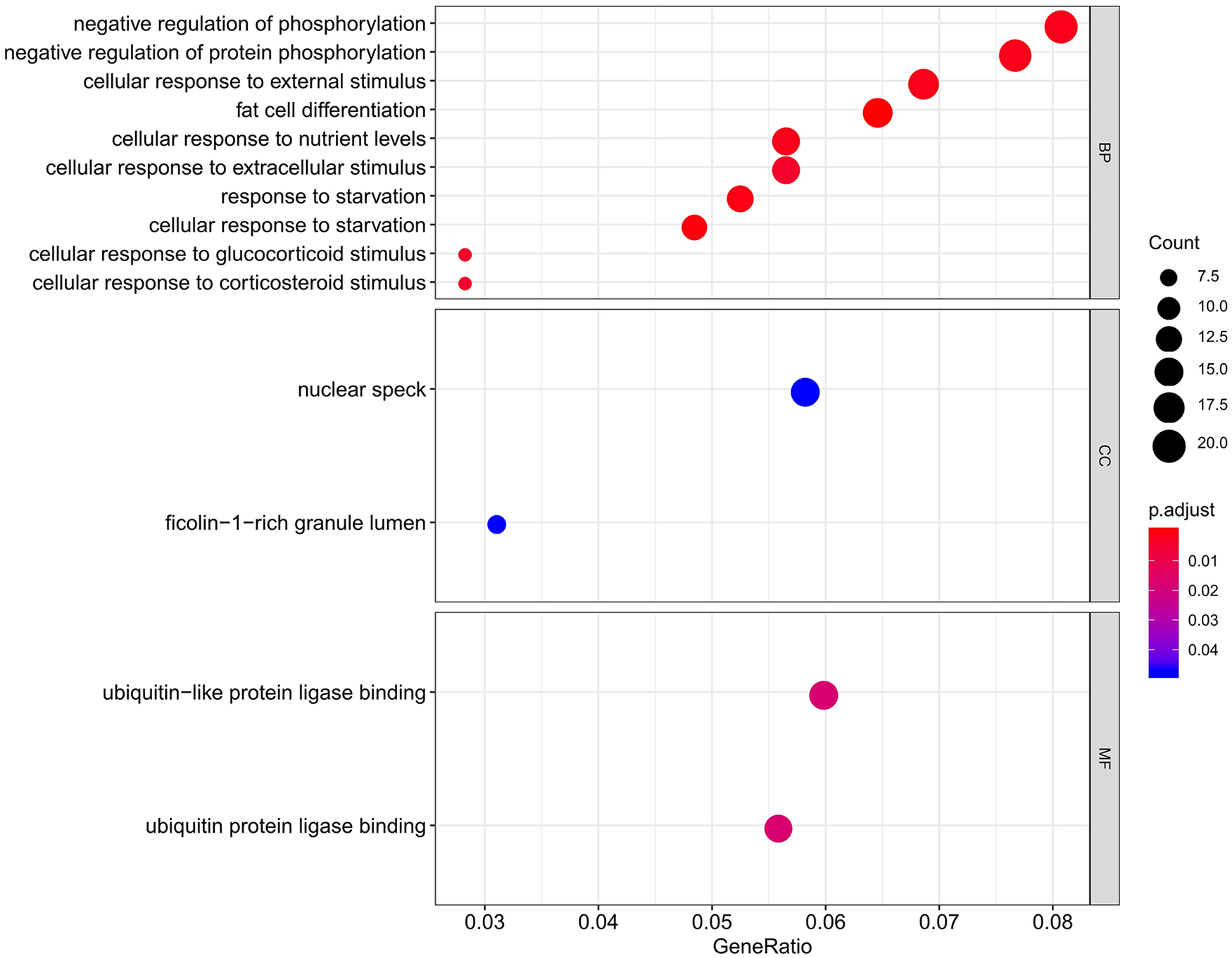
Gene Ontology (GO) enrichment analysis according to the differentially expressed genes. The gradual bubble color represents the adjusted P value and the increased bubble size represents the gene count.
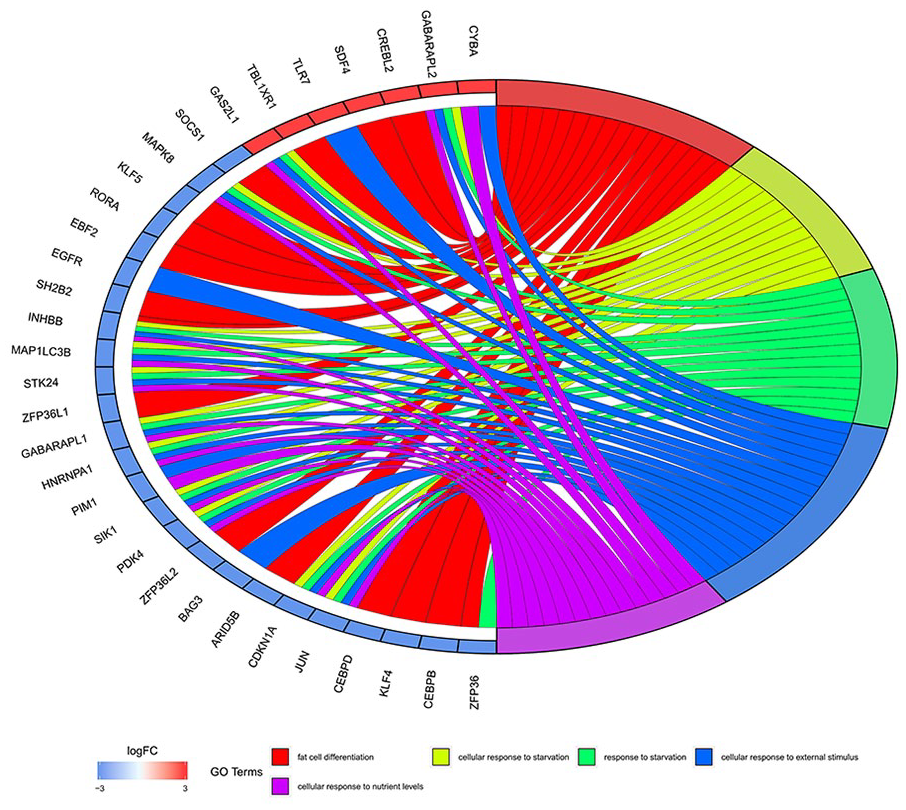
Top 5 key Gene Ontology (GO) terms tinvolved in the osteoarthritis (OA). For left semicircle, the gradual color represents log fold change (FC) of each gene. For right semicircle, different color represents the corresponding GO terms.
KEGG Pathways
KEGG pathways enrichment analysis revealed that DEGs between N and OA samples were involved in multiple pathways. Top 10 signaling pathways were FoxO signal pathway (hsa04068), IL-17 signaling pathway (hsa04657), Osteoclast differentiation (hsa04380), Breast cancer (hsa05224), Endocrine resistance (hsa01522), Melanoma (hsa05218), Epstein-Barr virus infection (hsa05169), parathyroid hormone synthesis, secretion, and action (hsa04928), autophagy-animal (hsa04140), and Salmonella infection (hsa05132). Top 10 KEGG enrichment pathways were shown in a bubble graph (
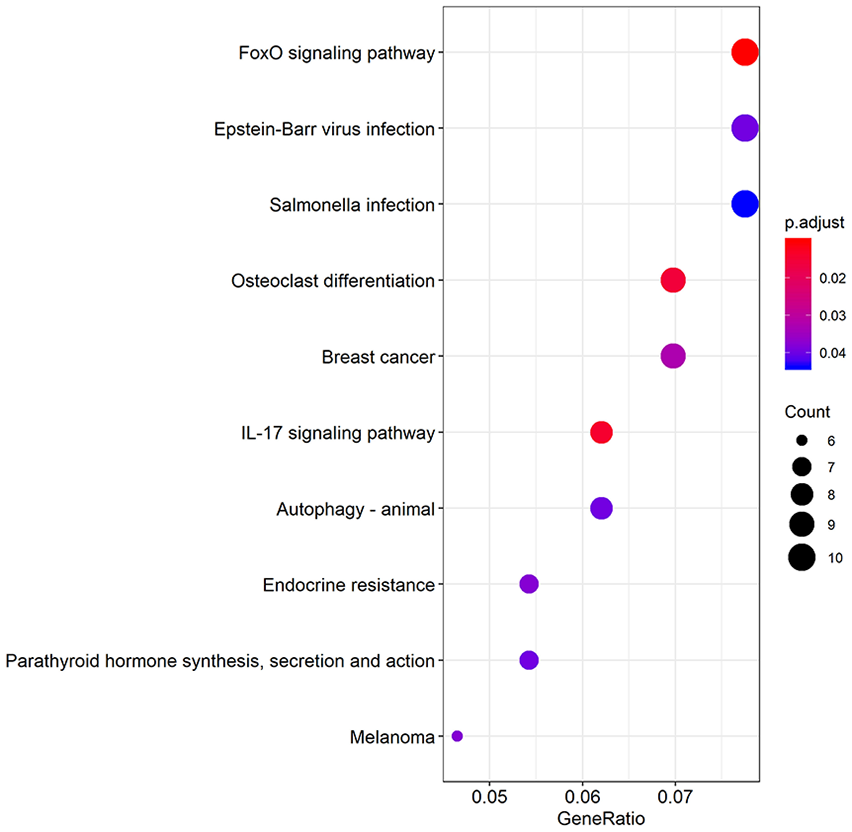
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis according to the differentially expressed genes. The gradual bubble color represents the adjusted P value and the increased bubble size represents the gene count.
PPI Network and Key Genes
PPI network analysis contained 259 interactive pairs and 133 nodes. Among these nodes, 47 genes were upregulated and 86 genes were downregulated. The network diagram revealed the connection among each protein (
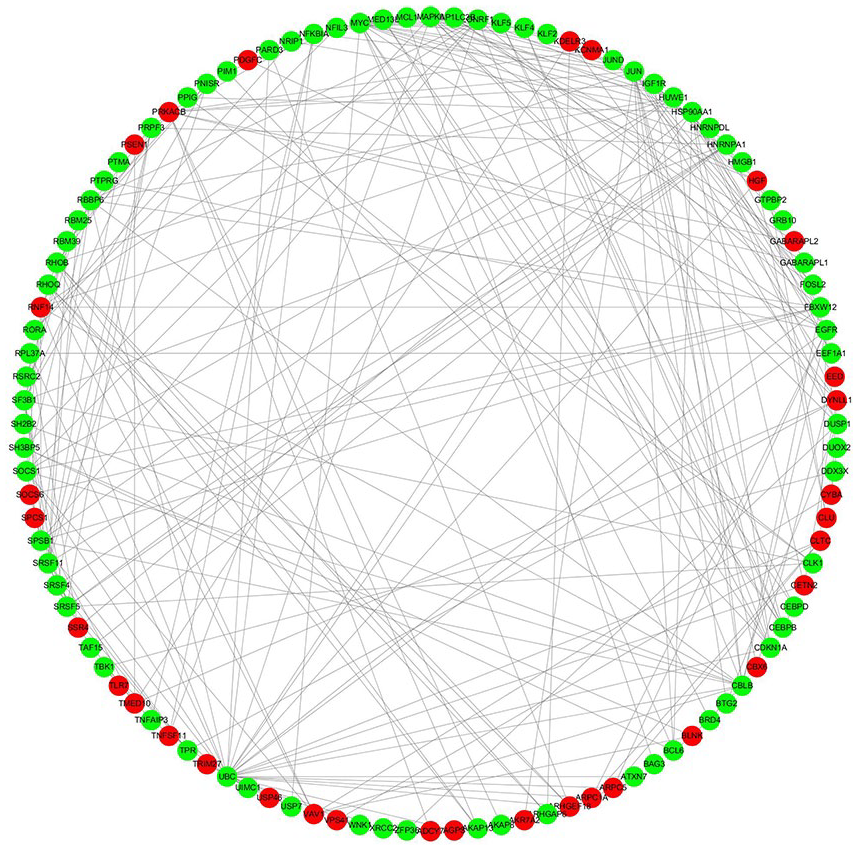
Protein and protein interaction (PPI) network according to the differentially expressed genes. The red circles represent the upregulated proteins and the green circles represent the downregulated proteins. The gray lines represent the interaction between 2 proteins.
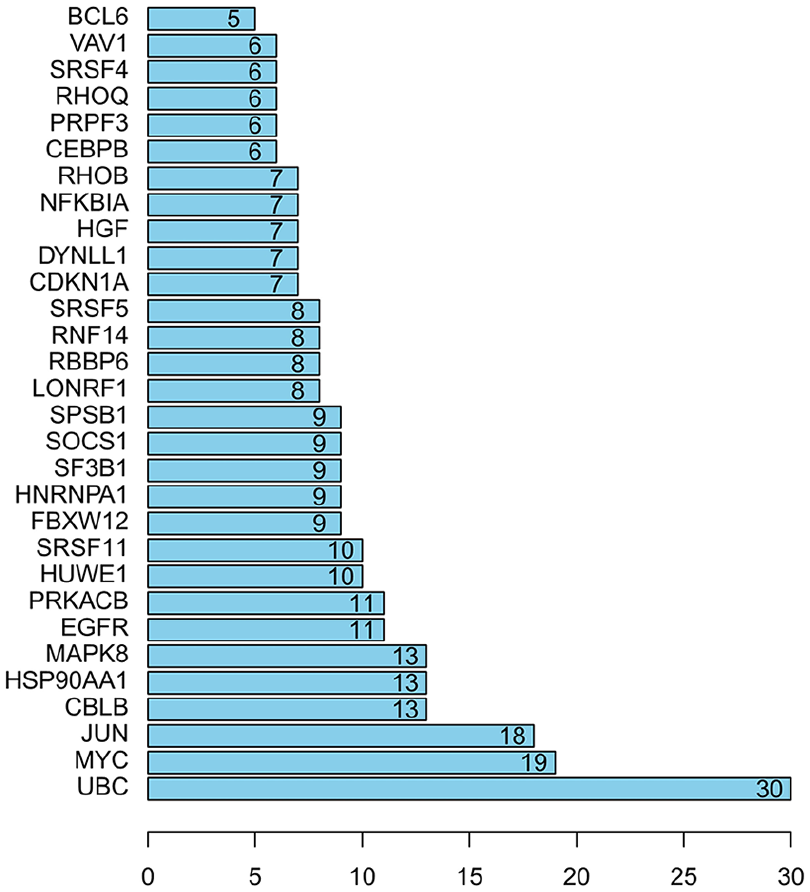
Top 20 key proteins involved in the OA. The horizontal axis represents the node degrees and the vertical axis represents the gene names.
Discussion
Based on the data of GEO database, the authors screened and selected 3 GEO series: GSE55235, GSE55457, and GSE82107 for analyzing the potential key genes involved in OA pathogenesis. After combination of each database, 274 DEGs were identified and some alternative genes for example BCL6, KLF9, and ZNF529 differed significantly in OA samples. According to GO analysis, cell differentiation, starvation, and negative regulation of phosphorylation were regarded as the most crucial biological process in the development of OA. Regarding the KEGG enrichment, FoxO and IL-17 signaling pathway along with osteoclast differentiation played important roles in OA. With regard to PPI network, UBC, MYC, and JUN generated most connections with other proteins and mainly participated in the OA progressing.
According to the screened result of DEGs, the authors disclosed many new different expressed genes in OA. BCL6 Transcription Repressor (BCL6) is a kind of transcriptional repressor that mainly interacted with immune cells like T-cell and B-cell. 9 It is reported that BCL6 is widely expressed in B-cell lymphomas such as large B-cell lymphoma (DLBCL) and follicular lymphomas (FLs), thus regulates the fate of multiple lymphomas. 10 BCL6 is also associated with T-cell by modulating the differentiation of CD4+ T follicular helper cells (Tfh).11,12 Considering the strong immunomodulatory effects acted by BCL6, we assumed that downregulated level of BCL6 may affect the microenvironment in knee joint especially for the synoviocytes. However, due to few studies reporting the function of BCL6 in OA, we will design further experiments to validate this hypothesis. Zinc Finger Protein 529 (ZNF529) is another hub gene infiltered by our study, which showed significant upregulation in OA. The structure of zinc finger is a small but functional domain that receive coordination of zinc ions.13,14 Through binding different nucleic acid, zinc finger proteins participate in varieties of biological functions. Previous study demonstrated that zinc finger, CCHC domain-containing protein 6 (ZCCHC6) aggravated severity of OA via increasing the expression of interleukin-6 (IL-6). Their mice experiment also verified the negative impact of ZCCHC6 in vivo. 15 However, it is worth noting that the regulations of zinc finger proteins are multidirectional; another study found that zinc finger protein A20 (ZFNA20) alleviated the inflammatory response by suppressing nuclear factor-kappa B (NF-κB). Thus, the complicated function of ZFN requires more exploration. 16
In the aspect of signaling pathway, Forkhead box O (FoxO) was detected as the most key pathway involved in OA. As a transcription factor, FoxO family including FoxO1, FoxO3, FoxO4, and FoxO6 regulate multiple biological processes: proliferation, differentiation, apoptosis, DNA repair, and so on. Utilizing the DNA binding domain (forkhead box), FoxOs combine with the cis-acting elements such as promoters, thus affecting the transcriptional activity of target genes. During the generation, development, and maturation of cartilage, FoxOs play an important role. Maladjustment of the FoxO pathway deteriorates the homeostasis of cartilage and synovium and contributes to pathogenesis of OA. A recent study reported that FoxO1 exhibited a salutary effect on chondrocytes by upregulating the expression of proteoglycan 4 (PRG4) and modulating cellular autophagy. 17 Another study illustrated that both FoxO1 and FoxO3 reduced markedly in meniscus from either human or mice OA samples, indicating the positive influence of FoxO pathway on supporting homeostasis and antagonizing degeneration.18,19 Following the FoxO, interleukin-17 (IL-17) was the other crucial pathway from our enrichment analysis. Although IL-17 increased more apparent in rheumatoid arthritis compared with OA, latest studies began to report the pivotal role of IL-17 in OA. 20 Upregulated IL-17 concentration in blood activates excessive inflammatory response and lead to perichondral damage and subchondral bone resorption. It is also proved that IL-17 activated autophagy, simultaneously impaired apoptosis, and result in destructive pathology of OA. 21 Moreover, IL-17 broke the balance of oxidative respiratory chain and caused the dysfunction of mitochondria. 22 Beyond to cartilage, more recent studies corroborate pathological change of subchondral bone can accelerate OA, in which abnormal osteoblast and osteoclast differentiation impair the bone homeostasis that is consistent with our enriched results.23,24 As one of the monocyte-macrophage systems, osteoclasts are mainly responsible for bone resorption to maintain the balance of bone metabolism. However, aberrant osteoclast activity destroys bone trabecular structure and results in excessive bone loss of subchondral bone at early-stage OA. 25 Our recent study found in in vitro OA environment, an osteoclast marker, matrix metalloproteinase 9 (MMP9) was increased in human chondrocytes. 26 Furthermore, hyperactivated osteoclastogenesis are also detected in in vivo models, indicating the key function of osteoclasts in OA development. 27
Taking the results from GO enrichment and PPI network, we discovered a critical molecular Ubiquitin C and its corresponding molecular function ubiquitin-like protein ligase binding and ubiquitin protein ligase binding. Ubiquitin-proteasome is thought to act as an indispensable recycling system that delivers substrate to proteasomes and lysosomes. 28 There is no doubt that ubiquitination participates in the degradation of chondrocytes, synoviocytes, and osteocytes. Ubiquitin-fold modifier 1 (UFM1)-specific ligase 1 (UFL1), which acts as an UFM1 E3 ligase, benefited the proliferation and differentiation of OA chondrocytes and inhibited the expression of several degrading enzymes by directing the NF-κB pathway. 29 Ubiquitin-specific protease 3 (USP3) prevented cellular apoptosis induced by IL-1β and regulated inflammatory and immunological response by interacting with tumor necrosis factor-receptor-associated factor 6 (TRAF6). 30 Unfortunately, no published study clearly elucidated the role of ubiquitin C in OA, hence more research on this molecule is needed. Regarding BP enrichment results, we noticed that negative regulation of phosphorylation is a vital biological process in OA. It is considered that as an epigenic regulation, phosphorylation and dephosphorylation are involved in numerous biologic behaviors. With the aspect of OA, activated phospho-nuclear factor-κB p65 (p-p65), phospho-phosphoinositide 3-kinase (p-PI3K) may aggravate OA by augmenting apoptosis while elevated phospho-AMP-activated protein kinase (p-AMPK) may protect OA via enhancing autophagy,31-33 suggesting the intervention of phosphorylation may be a candidate therapeutic target for OA in future.
The present bioinformatic analysis introduced some new insights based on the updated GEO datasets. DEGs including BCL6 and ZNF529, signaling pathway like FoxO, and key proteins containing UBC were identified and proposed for the first time. However, our study consisted of some limitations: (1) The enrolled samples from each GEO dataset were not large which may cause a degree of bias. (2) Our study was performed based on online databases and some results need further experiments to corroborate.
Conclusion
In summary, our study revealed 274 DEGs in OA pathogenesis using bioinformatic analysis. Hub genes like BCL6, ZNF529 and key signaling pathway FoxO were firstly uncovered by integrating high-throughput data. Our analyzed results identified valuable information about candidate genes, proteins, and pathways, which may provide new targets for OA therapy.
Supplemental Material
sj-pdf-1-car-10.1177_19476035211008975 – Supplemental material for Identification of Key Genes and Pathways in Osteoarthritis via Bioinformatic Tools: An Updated Analysis
Supplemental material, sj-pdf-1-car-10.1177_19476035211008975 for Identification of Key Genes and Pathways in Osteoarthritis via Bioinformatic Tools: An Updated Analysis by Yijian Zhang, Tianfeng Zhu, Fan He, Angela Carley Chen, Huilin Yang and Xuesong Zhu in CARTILAGE
Footnotes
Author Contributions
Yijian Zhang and Xuesong Zhu designed the research study; Yijian Zhang wrote the original article; Yijian Zhang, Fan He, and Angela Carley Chen performed the data analysis; Huilin Yang, Tianfeng Zhu, and Xuesong Zhu participated in the conception of the study and revision of the manuscript. All authors approve of the final version to be published.
Acknowledgements and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China [81772358, 82072442].
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The design of the present study adhered to the tenets of the Declaration of Helsinki.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.