
Abstract
Objective
Osteoarthritis (OA) is a chronic arthropathy that frequently occurs in the middle-aged and elderly population. The aim of this study was to investigate the molecular mechanism of OA based on autophagy theory.
Design
We downloaded the gene expression profile from the Gene Expression Omnibus repository. Differentially expressed genes (DEGs) related to the keyword “autophagy” were identified using the scanGEO online analysis tool. DEGs representing the same expression trend were screened using the MATCH function. Clinical synovial specimens were collected for identification, pathological diagnosis, hematoxylin and eosin staining, and real-time polymerase chain reaction analysis. Differential expression of mRNAs in the synovial membrane tissues and chondrocyte monolayer samples from OA patients was used to identify potential OA biomarkers. Protein-protein interactions were established by the STRING website and visualized with Cytoscape. Functional and pathway enrichment analyses were performed using the Metascape database.
Results
GABARAPL1, GABARAPL2, and ATG13 were obtained as co-expressed autogenes in the 3 data sets. They were all downregulated among OA synovial tissues compared with non-OA synovial tissues (P < 0.01). A protein-protein interaction network was constructed based on these 3 genes and included 63 genes. A functional analysis revealed that these genes were associated with autophagy-related functions. The top hub genes in the protein-protein interaction network were presented. Furthermore, 3 key modules were extracted to be core control modules.
Conclusions
These results offer an important molecular understanding of the key transcriptional regulatory genes and modules based on the network of potential autophagy mechanisms in human OA.
Background
Osteoarthritis (OA) is a chronic degenerative joint disease that causes loss of cartilage, inflammation of the synovial membrane, remodeling of subarticular bone, and formation of osteophytes. 1 This pathological process is an irreversible and progressive condition that affects more than 250 million people worldwide.2,3 Patients suffer from joint pain, stiffness, and swelling, which finally lead to tendon contractions, muscle spasms, and ligament laxity. Multiple risk factors contribute to the progression of OA, including genetic factors, aging, abnormal mechanical stress, and obesity. 4 A series of genes and signaling pathways have been identified to be correlated with the development of OA, 5 in which aberrant expression of inflammatory mediators, enzymes, and other proteins are involved, such as collagenases, nitric oxide synthase 2, cyclooxygenase 2, and serine proteinases. 6
Autophagy is part of the lysosomal degradation pathway essential for cell survival. Autophagy is a dynamic and sequential process in eukaryotic cells that principally involves induction, nucleation, elongation, maturation, fusion, and degradation. 7 In the growing cell, autophagy is always induced in response to cellular stress. It is maintained and serves as a quality control pathway by eliminating long-lived proteins and damaged organelles and recycling of cellular components. 8 In both its basal and induced states, autophagy is necessary for cell survival and maintaining and/or restoring homeostasis. The role of autophagy may vary with different stages and models of OA as it is recognized to be of cytoprotection to chondrocytes. Several knockout studies added evidences that dysregulated autophagy processes would be involved in the pathogenesis of OA.9-11 And the reducing expression of autophagy related factors was observed in OA or aging joints in humans and mice, accompanied by increased chondrocyte apoptosis. 12
Many autophagy-related mediators are present in OA. The functional relationship between autophagy and OA is quite complex. A large number of autophagy markers are correlated with OA pathology, including mTOR, proteoglycans, and Unc-51-like kinase 1,13,14 some of which have been identified as promising OA targets. Based on these findings, biological and pharmaceutical agents that regulate chondrocyte autophagy could be part of potential therapeutic methods.
To identify expression of these differentially expressed genes (DEGs) related to autophagy, OA and non-OA specimens were enrolled for experimental validation. Focusing on the autophagy-related mechanism contributing to OA, this study used a bioinformatics approach to construct a promising regulatory network to explore the autophagy-related modules. In addition, we distinguished pivotal genes and key cellular signaling pathways that may potentially contribute to the OA progression. This method will demonstrate the complete autophagy regulatory network and commence new fields of study in the pathophysiology of OA.
Methods
Microarray Data Collection and Screening
From the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), we searched and screened the microarray data set with the keyword “osteoarthritis,” following the essential points: (1) reliable source and detailed information; (2) expression profiling by array; and (3) more profiled samples in the database. Based on these instructions, GEO accessions GSE16464, GSE55235, and GSE55457 were selected for analysis.
We compared the OA monolayer chondrocytes versus the healthy joint expression data set (GEO accession GSE16464) and OA synovial tissue versus the healthy joint expression data sets (GEO accessions GSE55235 and GSE55457) in the GEO database. These 3 microarray data sets were, respectively, adopted to the following platforms: GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array, GPL96 (HG-U133A) Affymetrix Human Genome U133A Array, and GPL96 (HG-U133A) Affymetrix Human Genome U133A Array.
Data Processing
The 3 aforementioned data sets were chosen through a ScanGEO 15 (http://scangeo.dartmouth.edu/ScanGEO/) online R language analysis. ScanGEO rapidly mines high throughput gene expression data to identify genes and pathways of interest and answer biological questions relevant to diverse fields of study. Thus, the KEGG pathway was selected with the keyword “autophagy.” The original probe signal data in the microarray were corrected to obtain the log2 (IP/input) value. Expressions of genes related to the autophagy pathway were acquired by analysis of variance (P < 0.05) of the 3 data sets. DEGs were matched among the 3 microarray data sets, and significant genes were screened for further analysis.
Clinical Specimens and Experimental Validation
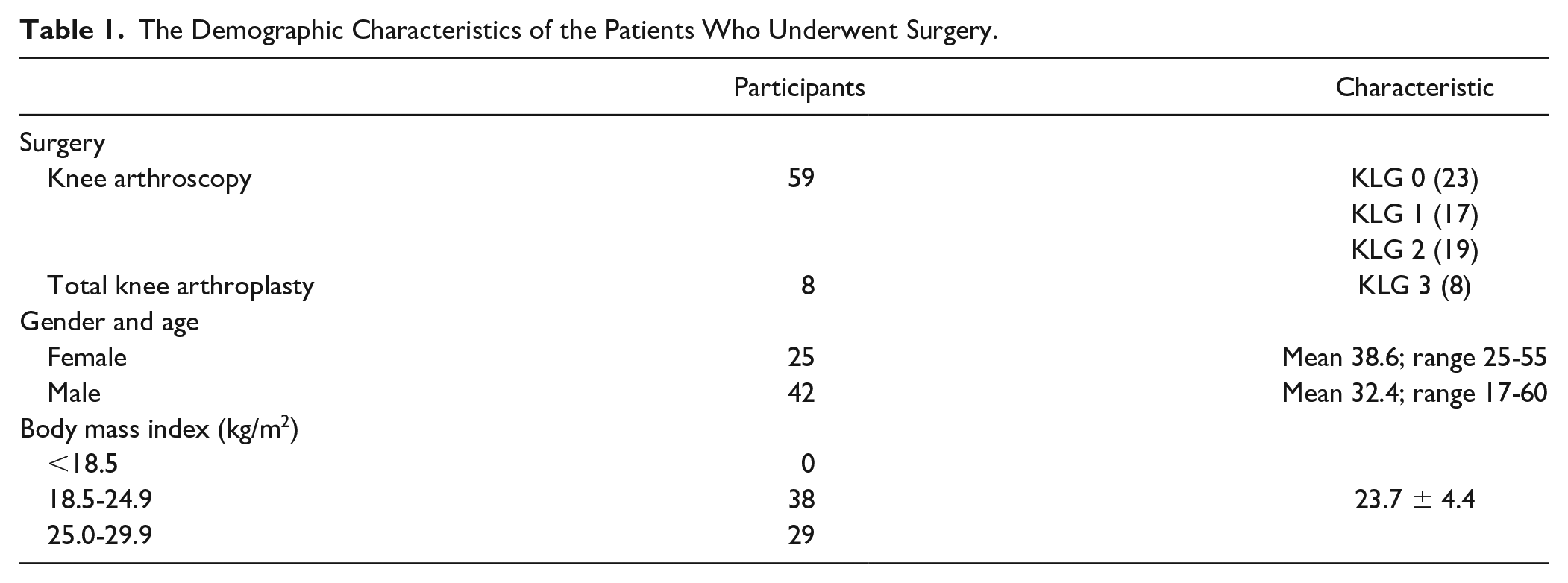
Synovial tissues were collected from 67 patients who underwent knee arthroscopy or total knee arthroplasty at Shengjing Hospital, China Medical University, from January 2017 to June 2019. The demographic characteristics of the patients who underwent surgery are listed in Table 1 . Pathologic diagnosis was ultimately provided by the pathologists. Hematoxylin and eosin (H&E) staining was carried out to observe the histopathological changes during the progression of OA. The real-time polymerase chain reaction (RT-PCR) was used to determine mRNA expression of GABA type A receptor associated protein like 1 and 2 (GABARAPL1, GABARAPL2), and ATG13. The primer sequences were the following: forward: 5′-GTGATTGTCCAGGCTCGGCTTG-3′ and reverse: 5′-TGTAACCTCTGGGATGTCTTTGATTGC-3′ for ATG13; forward: 5′-AGGGTGCCTGATCTGGACAAGAG-3′ and reverse: 5′-ACTGGTGGGAGGGATGGTGTTG-3′ for GABARAPL1; forward: 5′-CTGGAACACAGATGCGTGGAGTC-3′ and reverse: 5′-TCAATGTCAACAATCTGAGAGCCTGAG-3′ for GABARAPL2. All data were processed using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA) and expressed as mean ± standard deviation. Data were analyzed using a t test of 2 independent groups. A P value <0.05 was considered significant.
The Demographic Characteristics of the Patients Who Underwent Surgery.
Construction of the Protein-Protein Interaction Network
The protein-protein interaction network was mapped using the latest version of the STRING 11.0 (https://string-db.org/) online database, 16 which provides easy access to known and predicted protein interactions. After entering the significant genes from the previous analysis, STRING processes the data according to the following program: (1) existing experimental data mining, (2) computed prediction based on similar genes, (3) computer-based gene fusion, (4) protein homology, (5) other databases, and (6) gene co-expression networks. Interaction pairs with a confidence score >0.4 were inputted to construct the protein-protein interaction network. The results were calculated through the STRING database, which automatically generated and displayed the possible gene relationships. Furthermore, protein-protein interaction network was visualized by GENEMANIA (http://genemania.org/).
Enrichment Analysis
To find and confirm the major functions of the genes, we used Metascape (http://metascape.org) 17 to analyze the genes in the protein-protein interaction network by enrichment using the KEGG Pathway, GO Biological Processes, Reactome Pathway Database, Canonical Pathways, and CORUM.
Results
Identification of DEGs
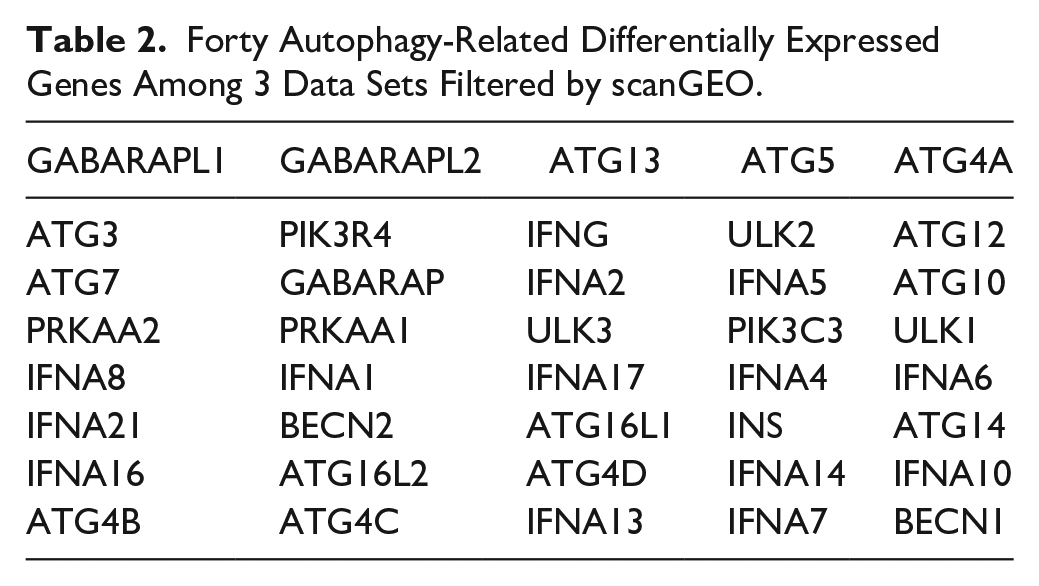
Three OA-related microarray data sets from Homo sapiens, such as GSE16464, GSE55235, and GSE55457, were screened. Using the scanGEO online R analytic process, the differential results of expression profiling by array were displayed under the condition of “autophagy,” including 40 DEGs ( Table 2 ) and 3 genes, such as GABARAPL1, GABARAPL2, and ATG13, obtained as co-expressed genes.
Forty Autophagy-Related Differentially Expressed Genes Among 3 Data Sets Filtered by scanGEO.
Histopathological Changes in the Specimens and Real-Time PCR Validation
Greater degrees of synovial hyperplasia, edema, fibrosis, and inflammatory cell infiltration were observed in the OA synovial group than in the normal synovial group in H&E staining ( Fig. 1 ). The OA progression of patients was consistent with their pathological diagnosis. Thus, the 67 specimens were divided into 2 groups of 23 normal specimens and 44 OA specimens. The real-time PCR analysis showed that GABARAPL1, GABARAPL2, and ATG13 was significantly downregulated in the OA group compared to the control group ( Fig. 2 ).
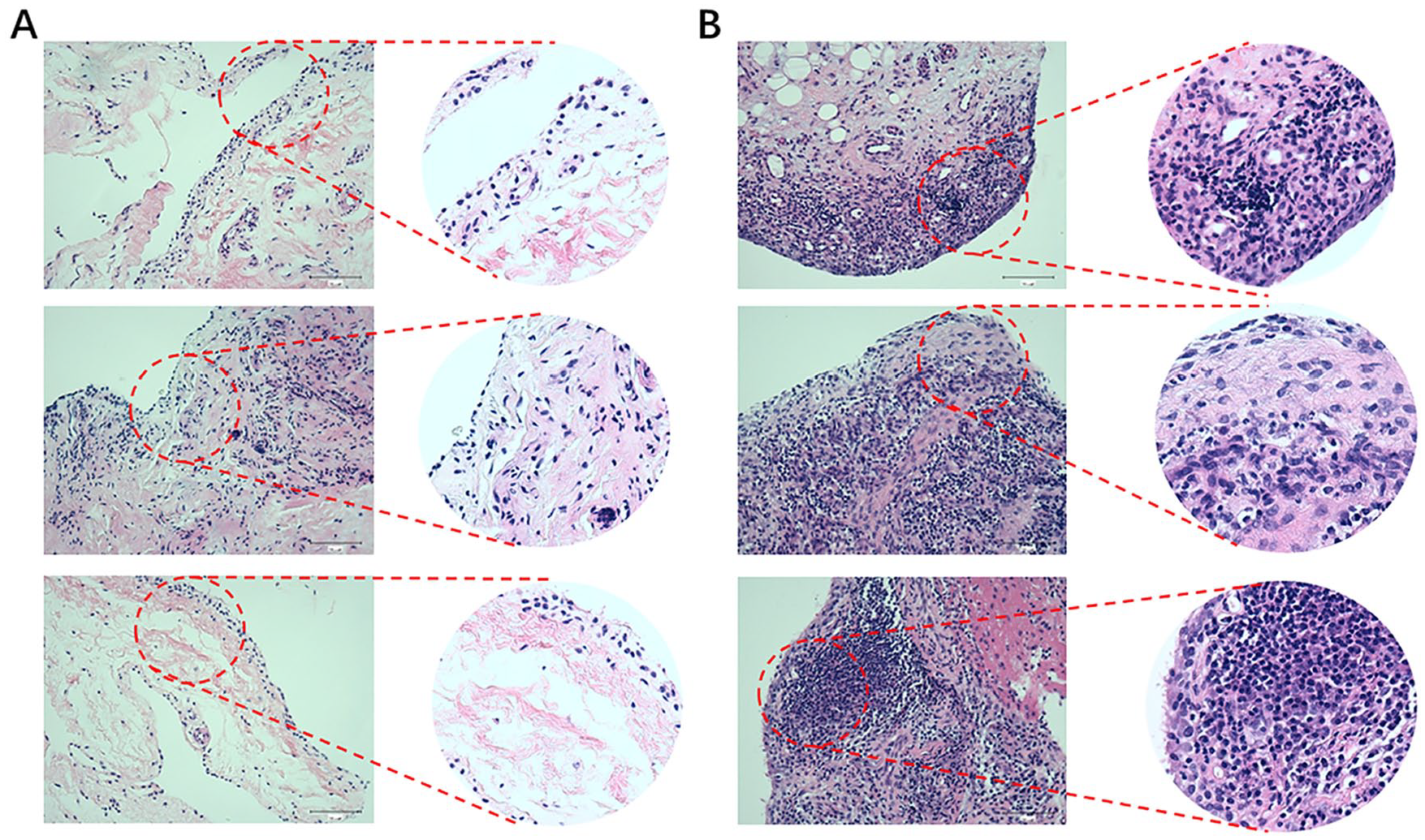
Hematoxylin-eosin staining and histological assessment of clinical human normal (
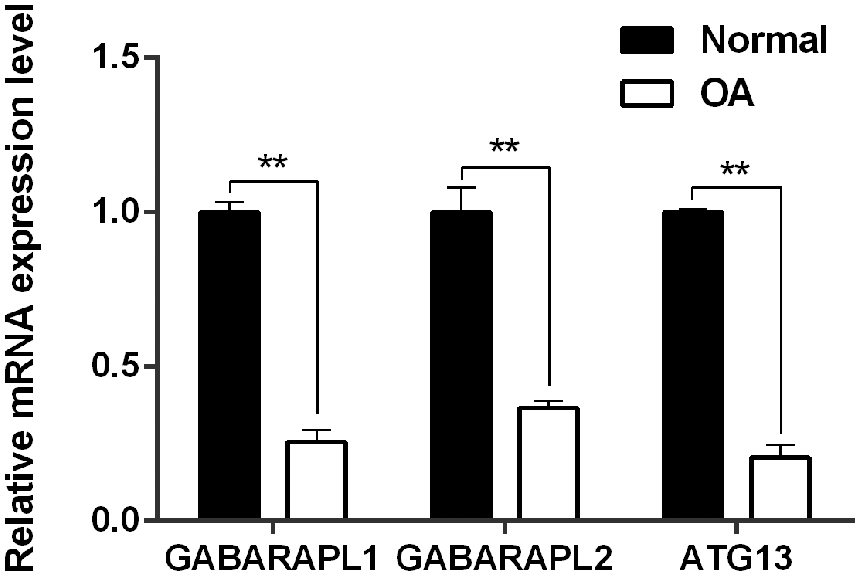
Relative mRNA expression level of GABARAPL1, GABARAPL2, and ATG13 between clinical normal and OA tissues. **P < 0.01 versus Normal synovial tissue.
Protein-Protein Interaction Network
Three key genes were integrated to develop the protein-protein interaction network including 63 genes ( Fig. 3 ). Seed genes were entered into the network and became the key nodes. Each node and its closest neighbor genes were extracted to construct a subnetwork of candidate genes. Sixty-three genes were obtained, and the calculated parameters were the following: number of nodes, 63; number of edges, 953; average node degree, 30.3; local clustering coefficient, 0.768; expected number of edges, 114; protein-protein interaction enrichment P value, <1.0e-16.
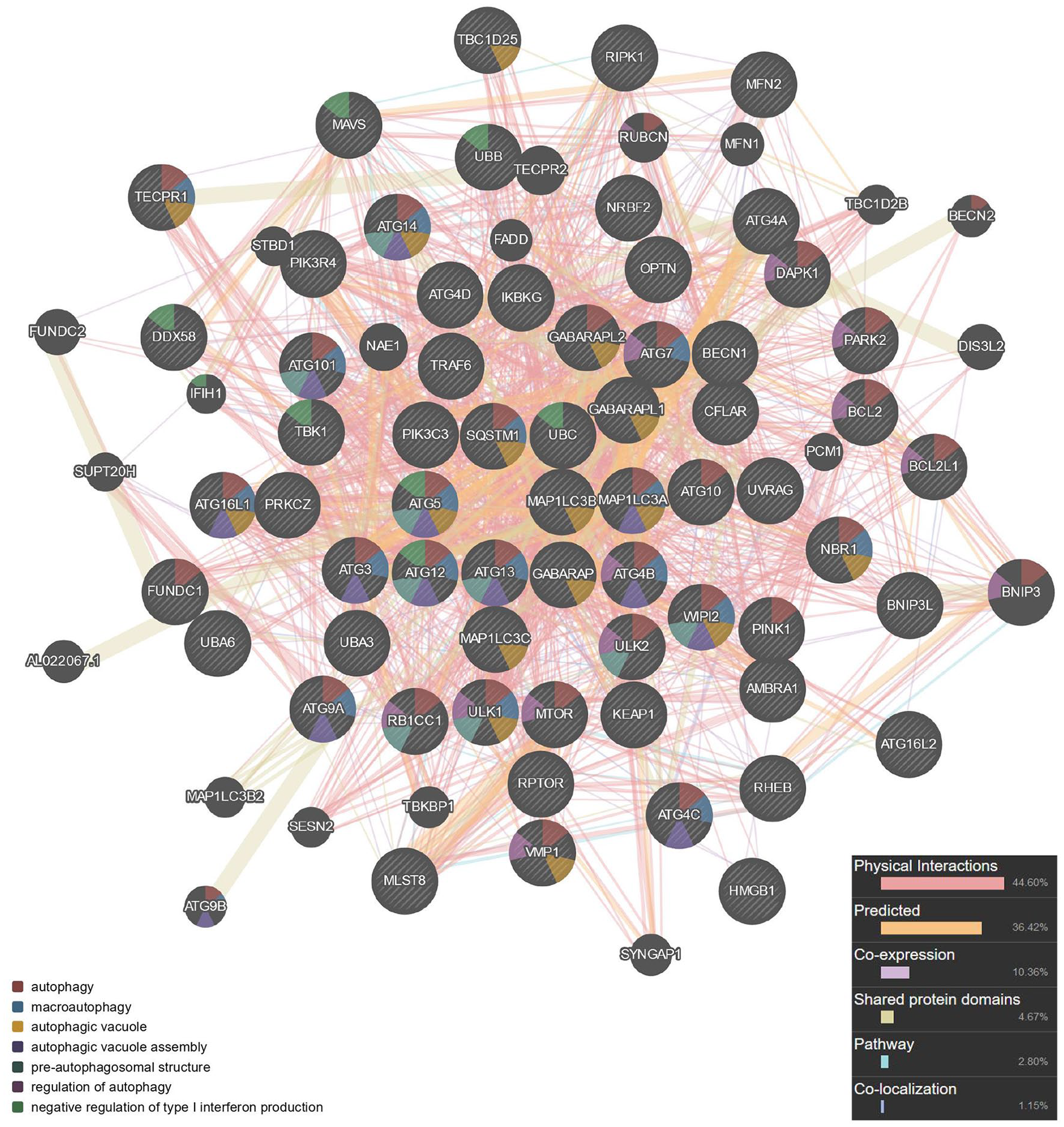
Protein-protein interaction network of autophagy construction.
Visualization of the Enrichment Networks
We implemented Metascape to visualize the pathway enrichment analysis. According to the results, 35 clusters with their enriched terms were shown, of which the top 20 are listed in Figure 4 . These clusters included genes with functions in macroautophagy, autophagy-animal, mitophagy-animal, selective autophagy, and response to starvation. The detailed information is listed in Table 3 . Enrichment networks were created by representing each enriched term as a node. Representative autophagy-related pathways were significantly enriched. Cytoscape provided visualization for the enriched term network in Figure 5 . The 63 genes associated with each pathway are shown in Supplementary Table 1. Three key modules, such as MCODE1, MCODE2, and MCODE3, are illustrated in red, blue, and green, respectively, using the MCODE function ( Fig. 6 ). MCODE1 included macroautophagy, autophagosome assembly, and autophagosome organization. MCODE2 included macroautophagy, autophagy-animal, and macroautophagy. MCODE3 included positive regulation of the response to a cytokine stimulus, DDX58/IFIH1-mediated induction of interferon-α/β (IFN-α/β) and negative regulators of DDX58/IFIH1 signaling.
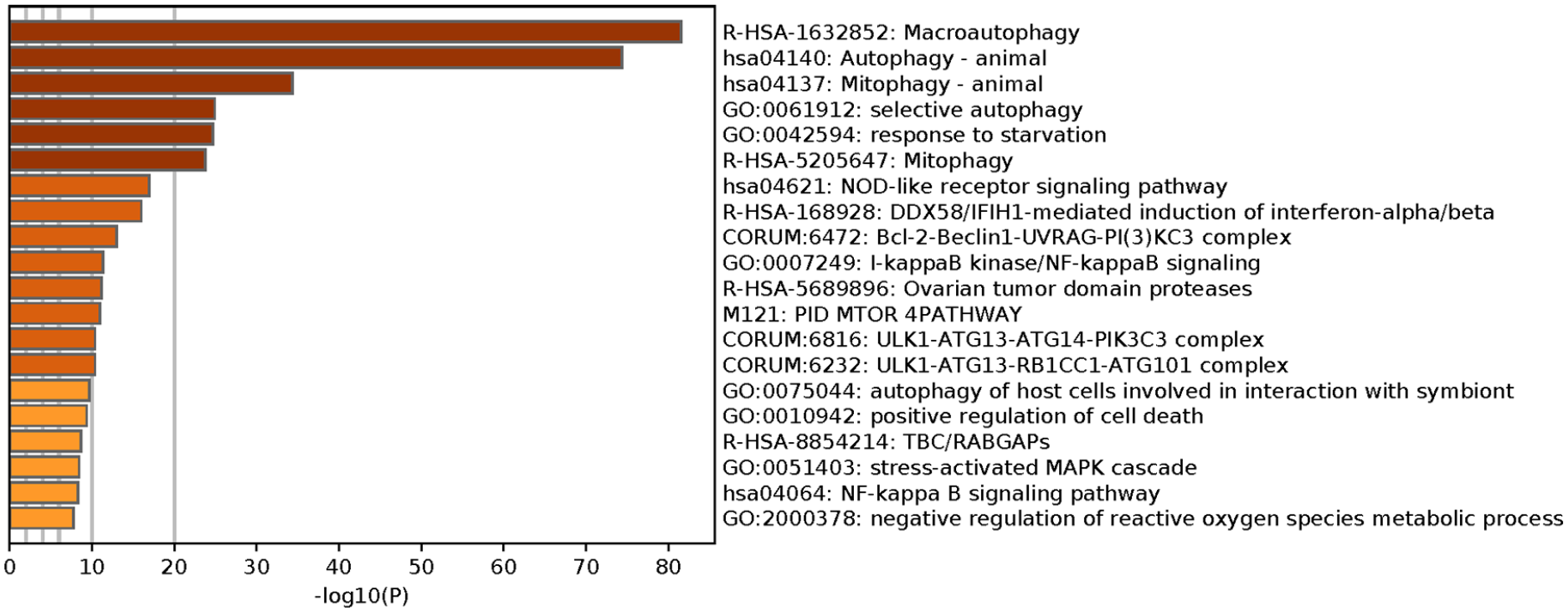
Metascape visualizes the top 20 clusters with their enriched terms and their related details.
Top 20 Clusters With Their Representative Enriched Terms (One per Cluster) a .
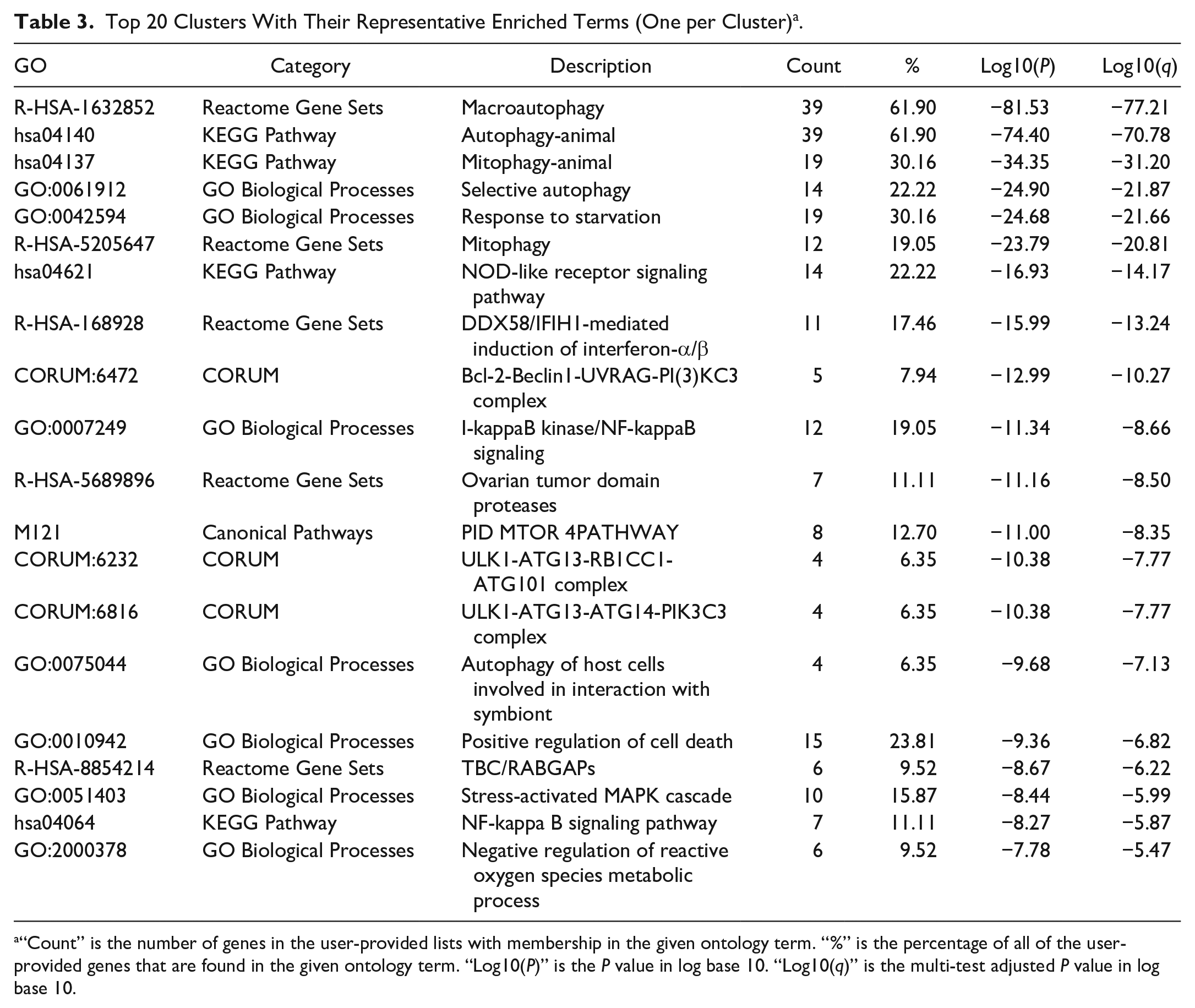
“Count” is the number of genes in the user-provided lists with membership in the given ontology term. “%” is the percentage of all of the user-provided genes that are found in the given ontology term. “Log10(P)” is the P value in log base 10. “Log10(q)” is the multi-test adjusted P value in log base 10.
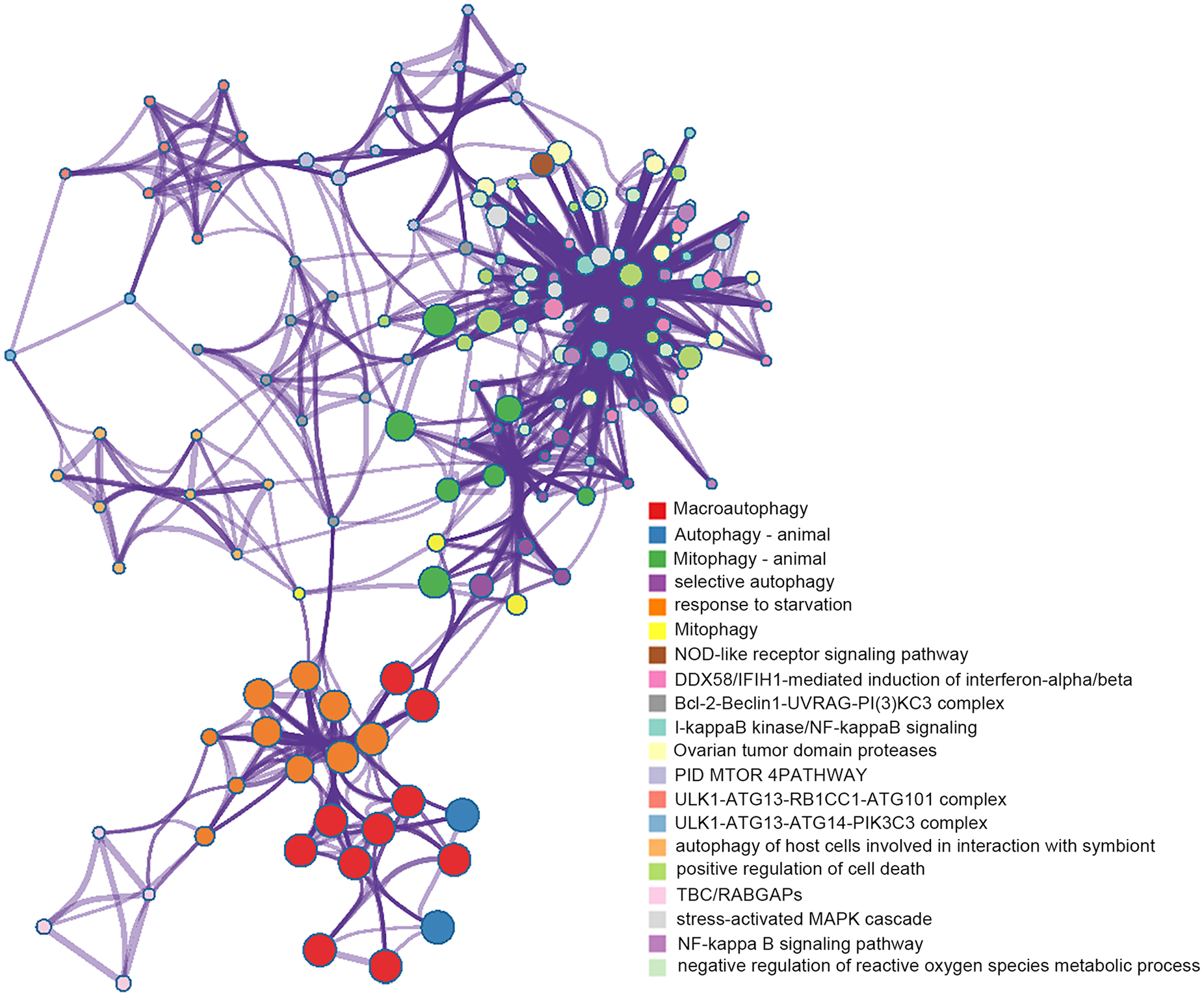
Visualization for network of enriched terms: colored by cluster ID, where nodes that share the same cluster ID are typically close to each other.
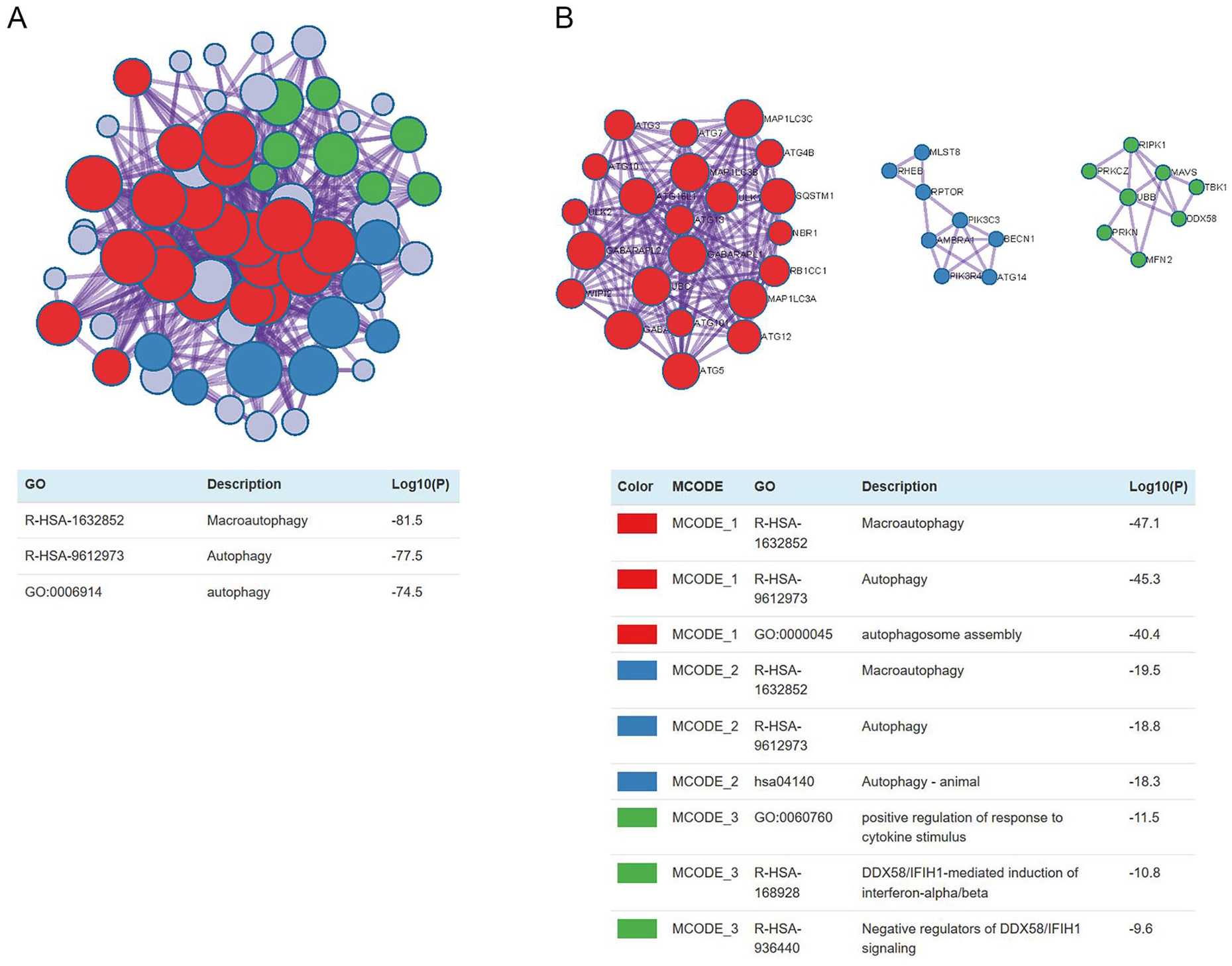
Protein-protein interaction network of hub genes and 3 MCODE components representing protein complexes embedded in the large network were extracted by MCODE function.
Discussion
Autophagy is a conserved maintenance mechanism in eukaryotic cells and is defined as a self-degradative nonapoptotic cell death process that is important for cartilage homeostasis. 18 Whereas in other cases it appears to be related to so-called autophagy-dependent cell death, defined as regulated cell death that depends on the autophagy machinery (i.e., pharmacological or genetic manipulations of autophagy genes block cell death), without involving alternative death pathways. The establishment of these criteria is critical, because autophagy is often observed in cell death scenarios, where it is activated in a failed effort to mitigate cell damage. In these latter cases, inhibition of autophagy promotes rather than protects from cell death. 8 Our discussion will be conducted in terms of its autophagy properties.
Dysfunction of chondrocyte autophagy is regarded as a pivotal pathogenesis of cartilage degradation in patients with OA. 7 Recent data suggest that enhanced autophagy flux could be protective during the early stages of OA, and is able to activate an adaptive response to cell stress to promote cell survival.19-21 Inhibiting autophagy attenuates the chondroprotective and anti-inflammatory effects. 22 The autophagy regulatory network exerts different mechanisms to balance homeostasis in the joints. However, knowledge about the mechanisms underpinning the occurrence and progression of OA in the elderly population is limited, particularly how autophagy is involved. Investigations into the autophagy regulatory network will significantly contribute to the diagnosis, treatment, and prognosis of patients suffering from OA. 23 High-throughput sequencing technologies and gene chips are postulated to detect the differential expression of genes in humans and have been a plausible strategy to study the molecular mechanisms of various diseases, predict potential therapeutic targets, and identify biomarkers. In this study, GEO accessions GSE16464, GSE55235, and GSE55457 were downloaded from the GEO database and a bioinformatics approach was applied to identify the key autophagy genes in OA.
Based on the screened autogenes, the overlapping autophagy-related DEGs in the 3 gene expression profile data sets were selected using the MATCH function, and the genes that were upregulated or downregulated were screened for further construction of the protein-protein interaction network. Only 3 key genes, such as GABARAPL1, GABARAPL2, and ATG13, were screened out as significant DEGs in the 3 data sets, indicating that these 3 genes were co-expressed autophagy-related genes in the progression of OA. GABARAP family have all been described to be composed of 3 members, GABARAP, GABARAPL1, and GABARAPL2. 24 GABARAPL1 and GABARAPL2 are defined as cognate autophagy proteins, 25 which closely interact with other autogenes.26,27 GABARAPL1 is able to increase basal autophagy flux independently of its conjugation to autophagosomes. 28 GABARAPL1 can also facilitate the fusion of autophagosomes with lysosomes. 29 GABARAPL2 plays a role in mitophagy, which contributes to regulating mitochondrial quantity and quality to a basal level to fulfill cellular energy requirements and preventing excess ROS production. 30 Autophagy-related (ATG) protein 13 (ATG13) is part of the ULK complex and the most upstream component in the autophagy pathway. 31 The initiation of cellular autophagy consists of a series of autophagy-related proteins such as Atg1 and Atg13. 32 Furthermore, the autophagy complex member Atg13 has been manifested in primary cultured osteoclasts and differentiation, which affects bone resorption.33,34
According to the imaging manifestations of the patients, the Kellgren-Lawrence Grade (KLG) was used to define the grade of OA. Twenty-three non-OA synovial tissues of knee trauma KLG0 patients were enrolled in the normal group. These patients underwent arthroscopic reconstruction of the cruciate ligament and repair of a meniscus injury; 44 patients were diagnosed with OA, including 17 KLG1 and 19 KLG2 patients who underwent arthroscopic joint cavity probing debridement, as well as 8 severe OA patients with KLG4 who underwent total knee arthroplasty. Patients were systematically diagnosed with or without OA according to clinical standards followed by corresponding surgical procedures. In addition, synovial histopathological results were verified by pathologists to definitively diagnose as the gold standard based at the histological level. The results indicated that OA progression was consistent with their pathological diagnosis. Thus, we defined OA synovial tissue versus non-OA synovial tissues for subsequent experimental verification. The histopathological analysis led us to conclude that the OA synovial tissues were more inflammation infiltrated than normal tissues. We confirmed in our study that mRNA expression of GABARAPL1, GABARAPL2, and ATG13 was downregulated in the OA samples. Thus, these 3 core autogenes might serve as significant mediators influencing the progression of OA.
We constructed a protein-protein interaction network using these 3 core autogenes. Sixty-three related genes were obtained and the protein-protein interactions illuminated the biochemical complexes or signal transduction components that govern biological outputs. 35 Some of these genes have been validated to be significantly altered in OA, such as BCL2L1, KEAP1, and RIPK1. Besides, 18 of these genes were mentioned in the genes that were previously screened to be differentially expressed in scanGEO, including ATG7, ATG12, ATG3, GABARAP, GABARAPL2, GABARAPL1, ATG16L1, ATG10, BECN1, ULK1, ATG14, PIK3R4, ATG13, ATG4B, ATG4D, ATG4A, ATG16L2, ATG4C. For example, some results suggested that ATG7 in human chondrocytes regulates autophagy by interacting with other mediators and it may become a more important target in OA progression or treatment.13,36,37 miR-128a-induced Atg12 loss repressed chondrocyte autophagy to aggravate OA progression. 38 Wu et al. pointed out that articular coinjection of bone marrow stromal cells-derived differentiated chondrocytes (DCs) and GABARAP could promote chondrocyte survival and slow OA progression. Mechanistically, GABARAP likely augments the protection effect of DCs in vivo by inducing autophagy and reducing the anti-inflammatory pathway. 39 The above information indicated that the PPI network built based on these 3 factors is meaningful.
These features were introduced to facilitate the interpretation of the enrichment analysis results obtained from Metascape. The Metascape results were dominated by macroautophagy, autophagy-animal, mitophagy-animal, selective autophagy, response to starvation, mitophagy, NOD-like receptor signaling pathway, DDX58/IFIH1-mediated induction of IFN-α/β, Bcl-2-Beclin1-UVRAG-PI(3)KC3 complex, I-kappa B kinase/NF-kappa B signaling, and positive regulation of cell death. Metascape applies a mature complex identification algorithm called MCODE to automatically extract protein complexes embedded in the large network and to infer more biologically interpretable results. This analysis helped identify groups or genes more closely related in the network. A detection (MCODE) algorithm has been applied to identify densely connected modules and network components. 40 Macroautophagy, autophagosome assembly, and autophagosome organization are directly correlated with the autophagy mechanism. Cytokine stimuli include many forms and are almost present in all cell metabolism. 41 Positive regulation of the response to a cytokine stimulus is closely correlated with autophagy, including nuclear factor-κB and matrix metalloproteinases in the development of OA.42,43 Macroautophagy/autophagy balances type I IFN signaling and plays essential roles in antiviral immunity.44,45 The neuroimmune axis is central in the physiopathology of OA, 46 and IFN is a major cytokine that regulates cellular immune reactions and serves as an important mediator in the pathogenesis of autoimmune diseases and participates in OA progression. 47 IFN-α/β belongs to Type I interferons and it has been implicated in the response of arthritic osteoblast to virus infection. 48 NOD-like receptor X1 (NLRX1) has emerged as an important regulator of inflammation and been reported to prevent in the pathology of OA. NLRX1 has been shown to negatively regulate Type I interferon, attenuate pro-inflammatory NF-kappa B signaling and modulate autophagy, cell death, and proliferation. 49 Furthermore, NLRX1 can also regulate NF-kappa B signaling, which indicates an anti-inflammatory role for NLRX1 in OA. 50 It is reported that the expression levels of various NOD-like receptors may determine their association with inflammatory cytokine levels in RA. 51 The relative amount of binding between bcl-2 and beclin 1 determines the cellular autophagy level and senescence. However, the regulatory mechanism for the autophagic degradation of DDX58 in OA remains largely undefined and is worthy of investigation. Application of autophagy on OA therapeutics will be possible after a profound understanding is established on the role of autophagy in OA pathogenesis.
Conclusion
Bioinformatics analysis is emerging as an efficient approach to identify key genes and molecular mechanisms. Taken together, the results of this study offer an important molecular understanding of the key identified genes and modules that may regulate transcription based on a network of autophagy potential mechanisms in human OA. However, prospective molecular experiments are needed to validate the function of these biomarkers associated with OA in the future.
Supplemental Material
Table_S1_Annotation_and_Enrichment_of_63_genes – Supplemental material for Transcriptional Regulation Based on Network of Autophagy Identifies KeyGenes and Potential Mechanisms inHuman Osteoarthritis
Supplemental material, Table_S1_Annotation_and_Enrichment_of_63_genes for Transcriptional Regulation Based on Network of Autophagy Identifies KeyGenes and Potential Mechanisms inHuman Osteoarthritis by Jiamei Liu, Qin Fu and Shengye Liu in CARTILAGE
Footnotes
Supplemental Material
Author Contributions
QF reviewed relevant literature and drafted the manuscript. JML and SYL conducted all statistical analyses, and the manuscript was approved by all authors for publication.
Authors’ Note
The data used and analyzed during the current study are available from GEO public database.
Acknowledgment and Funding
The authors want to show gratitude to the professors in the department who provided ideas for improvement in this project. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Outstanding Scientific Fund of Shengjing Hospital (MD31) and the National Natural Science Foundation of China (No. 81370981).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval was not necessary in our study because data are from a public database and experimental synovial tissues were discarded followed by pathologic diagnosis after surgery process.
Informed Consent
Verbal informed consent was obtained from all subjects before the study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.