
Abstract
Nicotinamide phosphoribosyltransferase (Nampt) catalyzes the rate-limiting step of nicotinamide adenine dinucleotide (NAD) synthesis. Both intracellular and extracellular Nampt (iNampt and eNampt) levels are increased in several human malignancies and some studies demonstrate increased iNampt in more aggressive/invasive tumors and in tumor metastases. Several different molecular targets have been identified that promote carcinogenesis following iNampt overexpression, including SirT1, CtBP, and PARP-1. Additionally, eNampt is elevated in several human cancers and is often associated with a higher tumor stage and worse prognoses. Here we review the roles of Nampt in malignancy, some of the known mechanisms by which it promotes carcinogenesis, and discuss the possibility of employing Nampt inhibitors in cancer treatment.
Introduction
Nicotinamide adenine dinucleotide (NAD) was first discovered in 1904 by Sir Arther Harden who identified a low-molecular-weight compound in yeast termed cozymase that was required for sugar fermentation. In the 1930s, Warburg found this compound to be a hydride-accepting and donating molecule that played a role in multiple cellular reactions. 1 The enzymatic activity responsible for NAD synthesis was identified in 1957 and designated as nicotinamide mononucleotide (NMN) pyrophosphorylase. 2 In 1966, Gholson predicted that NAD is actively turned over within cells, and this prediction was confirmed when the half-life of cellular NAD was found to be 1.0 ± 0.3 hours within cultured cells.3,4 The enzyme responsible for NAD synthesis was first cloned from activated peripheral human lymphocytes in 1994. It was initially identified as a secreted cytokine that synergized with interleukin-7 and stem cell factor to stimulate early stage B-cells, hence its designation as pre-B cell colony-enhancing factor (PBEF). 5 Later work confirmed a role for PBEF as a cytokine that is up-regulated in activated neutrophils and recombinant PBEF inhibits neutrophil apoptosis when placed in culture media. 6 In 2005, Fukuhara et al. 7 identified a visceral fat-secreted adipokine, corresponding to PBEF, with insulin-mimetic effects that was designated visfatin. The article was later retracted based on difficulty with data reproducibility. 8
Data demonstrating that PBEF played a role in intracellular NAD synthesis came in 2001, when Martin et al. 9 demonstrated that the Haemophilus ducreyi gene nadV, which has homology to mammalian Nampt/PBEF, is an NAD phosphoribosyltransferase that when expressed allowed H. ducreyi to grow in NAD free media. Later, Rongvaux et al. 10 demonstrated that murine PBEF was an intracellular nicotinamide phosphosribosyltransferase. The cloned murine Nampt gene was also able to confer growth in Actinobacillus pleuropneumoniae lacking nadV, indicating conservation of the NAD synthesis pathway between mammals and bacteria. 10 Based on this work, the enzyme was designated nicotinamide phosphosribosyltransferase (Nampt).9,10
Nampt is now known to catalyze NAD synthesis by transferring the phosphoribosyl group of 5-phosporibosyl-1-pyrophosphate to nicotinamide (NAM), forming NMN. NAD synthesis is completed by NMN adenylyltransferase (Nmnat), which converts NMN into NAD. 10 Nampt’s catalytic activity is ~46-fold lower than Nmnat activity, so Nampt catalyses the rate-limiting step of NAD synthesis. Thus, even very small changes in Nampt, but not Nmnat levels, can profoundly affect NAD metabolism and NAD-dependent events.10,11 The specific murine Nmnat isoform analyzed in this study was not identified, although the Nmnat had Km and Vmax values consistent with those previously reported for human Nmnat-1. 11 Interestingly, there are 3 different Nmnat enzymes located in the nucleus, cytosol, and mitochondria (Nmnats1-3, respectively).10,11 NAD is synthesized either de novo from precursors such as tryptophan, and nicotinic or quinolinic acids, or by the Nampt/Nmnat-catalyzed salvage pathway, with the later pathway being significantly faster, more efficient, and also the major NAD biosynthesis pathway in mammals (Figure 1).1,11,12 Nampt is found both intracellularly in the cytoplasm (designated iNampt), nucleus, and possibly the mitochondria in most cell types, and extracellularly in the plasma (eNampt), which was previously designated as visfatin.7,8,13,14
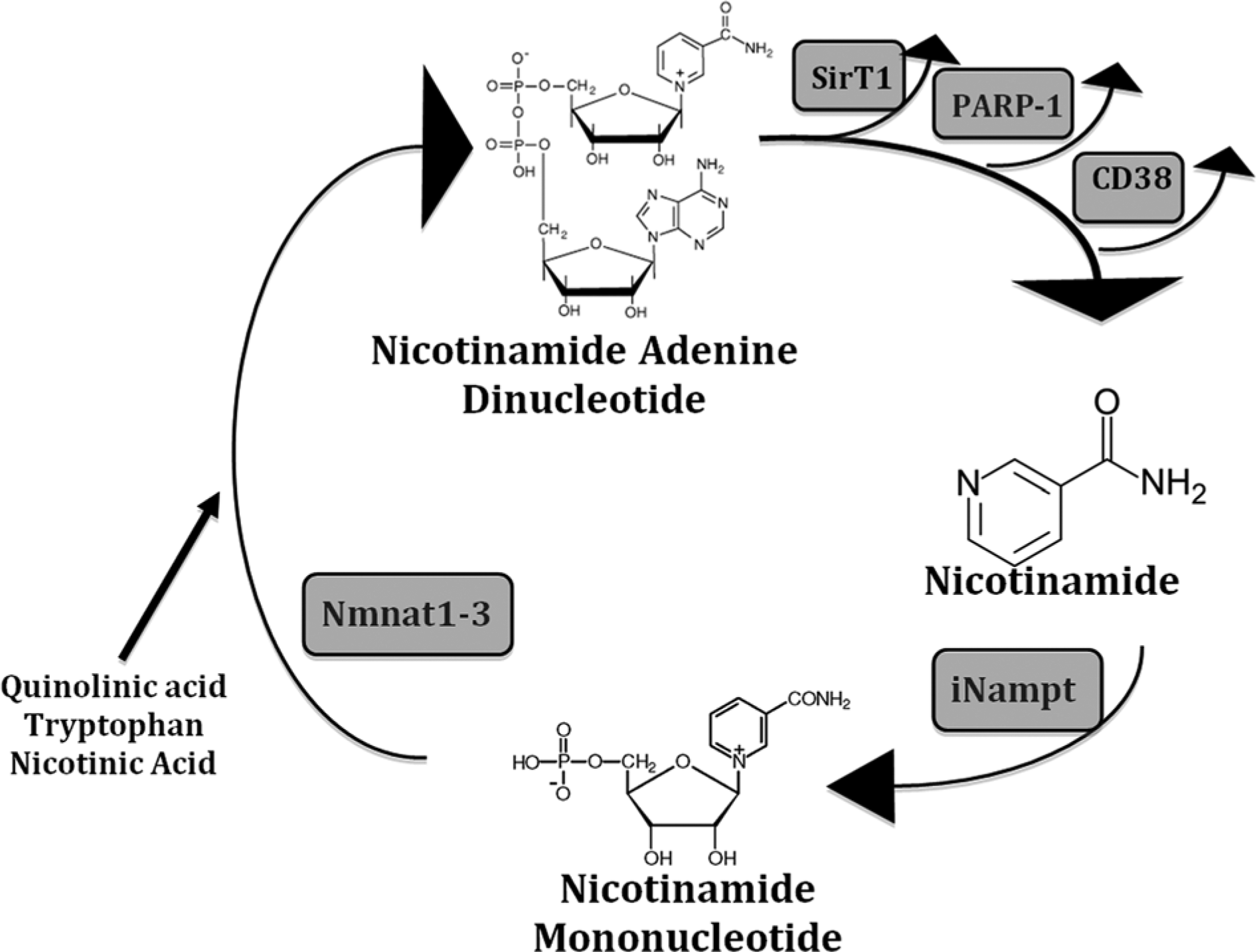
The NAD salvage pathway (modified from 11). NAD is consumed in many different reactions, including PARP1, CD38, and SirT1 activities.
Yang et al. 15 found that cell survival following genotoxic stress was dependent on the mitochondrial, but not the nuclear or cytoplasmic NAD pools. Cell survival was increased with increasing iNampt expression, an event dependent on expression of the mitochondrial NAD+-dependent deacetylases SirT3 and SirT4, indicating that mitochondrial Nampt/NAD synthesis plays a vital role in cellular function and survival. However, other investigators have not found mitochondrial Nampt. For example, Nikiforov et al. 16 found no mitochondrial iNampt and instead found that the mitochondrial-specific Nmnat3 produced all the mitochondrial NAD from NMN imported from the cytosol. The reason for this discrepancy is unknown, but it may be due to using different cells, or different mitochondrial isolation and analysis techniques. Interestingly, approximately 70% of the intracellular NAD pool is mitochondrial. 1
The functions of eNampt are presently poorly understood and there is conflicting data on eNampt function. For example, Revollo et al. 17 found that Nampt haplodeficient mice have reduced plasma NMN and eNampt and defects in glucose-stimulated insulin secretion in pancreatic β-cells. This defect is corrected by NMN administration. The authors concluded that eNampt-mediated systemic NAD synthesis is critical for normal β-cell function. However, Zhang et al. 14 found that eNampt promoted macrophage survival following endoplasmic reticulum stress, by stimulating interleukin-6 secretion and Stat3 activation. Interestingly, enzymatically inactive mutated eNampt was as biologically active as wild-type eNampt. Thus, eNampt exerts some functions independently of NAD synthesis. Last, low plasma eNampt levels correlate with hepatic mitochondrial dysfunction, thereby indicating that eNampt regulates some aspects of intracellular biochemistry. 18 An NAD-synthesizing role for eNampt also seems unlikely as other investigators have found eNampt to have very low catalytic activity under normal physiologic conditions, due to the extracellular space having very low adenosine-triphosphate concentrations.19,20 eNampt is secreted by a nonclassical pathway in several different cell types, including differentiated adipocytes, macrophages, cardiomyocytes, and hepatocytes.21 -25 Interestingly, overexpression and secretion of eNampt in murine cardiomyocytes resulted in cardiac hypertrophy. Cardiomyocyte eNampt secretion was inhibited in cell culture by treatment with NAM or trichostatin, indicating that extracellular signals my partially regulate eNampt secretion. 25
Nampt is found at 7q22 and spans 34.7 kb having 11 exons and 10 introns, giving a cDNA of 2,357 kb translated into a 491 amino acid, 52 kDa protein. Three predominant mRNA transcripts have been identified, comprising 2.0, 2.4, and 4.0 kb transcripts.13,15 Nampt mRNA is found in all tissues, suggesting it has a vital and indispensible function.10,26 The enzyme shows a high degree of evolutionary conservation, and enzymes with closely related sequences are found in prokaryotes, sponges, insects, and mammals.10,27 Crystallographic studies show that Nampt is a dimeric class type II phosphoribosyltransferases where 2 identical Nampt subunits contribute to an enzymatic active site, thus converting NAM and 5-phosporibosy1-1-pyrophosphate into NMN by an ANDN mechanism.27,28 iNampt is phosphorylated at histidine 247, resulting in a 160,000-fold increased enzymatic affinity for NAM. 29 iNampt and eNampt undergo other posttranslational modifications, including acetylation and ubiquitization. The significance of these modifications is presently poorly understood. 11 Presently all 3 of these names (PBEF, Visfatin, and Nampt) are used, although the Human Genome Organization Gene Nomenclature Committee approved the name of Nampt.
Nampt, NAD+, and Cancer
NAD is a cofactor that plays a central role in cellular electron transfer redox reactions, alternating between oxidized and reduced forms (NAD+ + e− ⇔ NADH) and is a universal energy- and signal-carrying molecule. NAD functions in many cellular events, including transcriptional regulation, longevity and caloric-restriction responses, cell cycle progression, apoptosis, DNA repair, circadian rhythms, chromatin dynamics regulation, telomerase activity, intracellular calcium mobilization. It also regulates the histone deacetylases (SirT1-T7), CtBP, CD38, and the poly(ADP-ribose) polymerases that play a central role in the maintenance of organismal metabolic homeostasis and genomic stability.1,30 -36 Unlike many cellular redox reactions, several NAD-dependent signaling processes degrade NAD by transferring the ADP-ribose moiety onto a receptor with the concomitant release of NAM. Thus, constant NAD resynthesis is an absolute requirement for cell survival, especially for rapidly growing cells. 33
Several different human malignant tumors have been demonstrated to overexpress iNampt including colorectal, ovarian, breast, gastric, prostrate, well-differentiated thyroid, and endometrial carcinomas, and myeloma, melanoma, and astrocytomas. Increased iNampt expression also occurs in malignant lymphomas, including diffuse large B-cell lymphoma, follicular B-cell lymphoma, Hodgkin’s lymphoma, and peripheral T-cell lymphoma (Table 1).37-55 eNampt also increases the growth fraction of the hepatocellular carcinoma HepG2 cell line in vitro, suggesting a role for it in hepatocellular carcinoma. 53 Many of these studies have found interesting aspects of Nampt expression in cancer. Olesen et al. 55 found higher iNampt expression in aggressive malignant lymphomas. Huang et al. 56 found that iNampt expression increases cellular stromal cell-derived factor-1 levels in colon cancer cells, promoting colorectal carcinoma progression. Although most studies documented increased iNampt levels between benign and malignant tissue, several correlated iNampt expression with specific changes in tumor behavior. For example, Long et al. 44 found iNampt was expressed 13 times higher in gastric cancer than in benign gastric tissue. Higher iNampt expression correlated with deeper tumor invasion, lymph node metastases, a higher clinical TMN stage, and a reduced patient survival. Similarly, increased iNampt correlates with increased tumor growth, metastases, cellular dedifferentiation, and the presence of a vertical growth phase in melanoma. Higher iNampt expression also confers a worse prognosis in endometrial adenocarcinoma and astrocytomas.47 -49,52 Wang et al. 45 found that elevated iNampt expression in early prostate cancer and inhibition of iNampt suppressed cell growth in culture, cell invasion, and the growth of xenografted prostate cancer cells in mice. Last, several researchers found that iNampt expression confers resistance to chemotherapeutic agents, including fluorouracil, doxorubicin, paclitaxel, etoposide, and phenylethyl isothiocyanate.40,41,43,45 Interestingly, in one case of signet ring cell gastric carcinoma following exposure to the Chernobyl nuclear accident, iNampt expression was very low in the malignant cells, which may suggest that iNampt expression may be different between radiation-induced and sporadic gastric cancers. 57
Nampt Overexpression in Several Human Malignancies a .
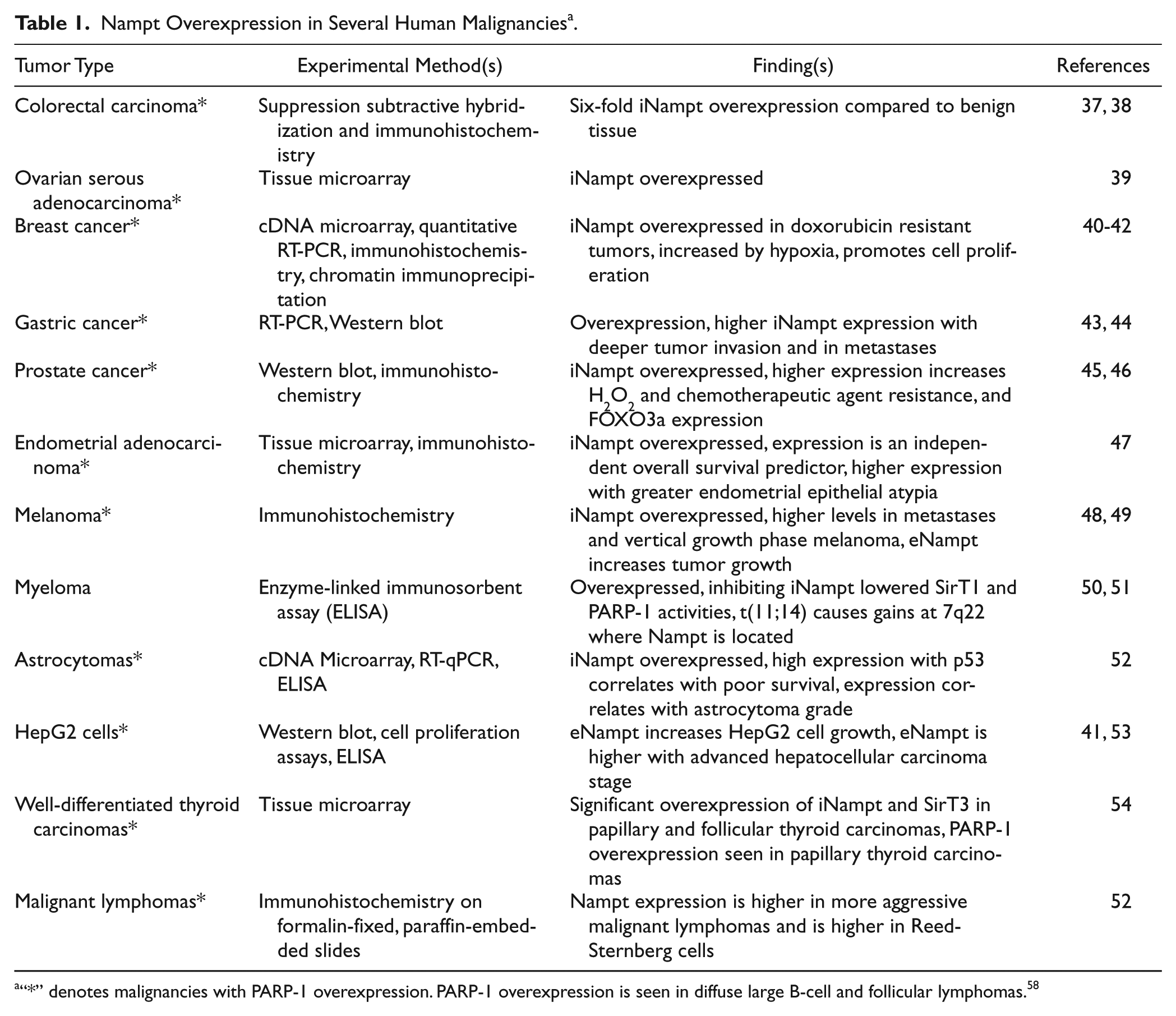
“*” denotes malignancies with PARP-1 overexpression. PARP-1 overexpression is seen in diffuse large B-cell and follicular lymphomas. 58
Molecular Mechanisms of iNampt and Carcinogenesis
Several molecular targets of iNampt activity have been identified that contribute, or are likely to contribute, to carcinogenesis and cancer progression. Most appear to be regulated by increased intracellular NAD synthesis and degradation. Here we will review several of them. The role of eNampt in carcinogenesis is discussed in a separate section.
SirT1
The silent mating type information regulation 1 (SirT1) is a sirtuin family member, which consists of 7 isoforms, each of which has specific functions and subcellular localizations. SirT1 is the best characterized of the sirtuins and functions as a longevity-promoting protein playing a role lifespan extension induced by caloric restriction. SirT1 is an NAD+-dependent histone deacetylase overexpressed in prostate, colon, breast, gastric, liver, and pancreatic tumors; interestingly, many of the tumors overexpress iNampt (Table 1). 59 Up-regulation of SirT1 in malignancies is associated with a poor prognosis, poor therapy response, shorter patient survival, higher tumor stage, node metastases, and increased Ki-67 expression. The molecular mechanisms of SirT1 function in cancer are complex, with some pathways promoting carcinogenesis and others suppressing it. For example, SirT1 activity suppresses Stat3 and NF-κB signaling and attenuates chronic inflammatory responses, suppressing carcinogenesis. However, SirT1 also attenuates p53, PTEN, retinoblastoma protein activities, stabilizes N-Myc, promotes the epithelial to mesenchymal transition, and increases cell migration, all of which promote carcinogenesis. 56 Revollo et al. 11 found that increased iNampt, but not Nmnat, increased cellular NAD levels, enhancing SirT1-mediated transcription in murine cells. Additionally, oligonucleotide microarray studies demonstrated a significant correlation in gene expression profiles of iNampt and SirT1 overexpressing cells. The authors concluded that iNampt-mediated NAD synthesis regulates SirT1 function. Not surprisingly, iNampt-mediated cellular resistance to oxidants is attenuated with SirT1 knockdown in prostate cancer. 45 Last, the mitochondrial-specific sirtuins SirT3 and SirT4 may function with mitochondrial Nampt to promote cell survival following genotoxic stress. 15 The significance of these sirtuins in cancer is so far poorly characterized, although interestingly both iNampt and SirT3 are overexpressed in well-differentiated thyroid carcinomas.54,60,61
CtBP
The mammalian COOH-terminal binding proteins (CtBPs), CtBP1 and CtBP2, promote invasive behavior and apoptosis resistance in malignant cells, with concomitant suppression of the tumor suppressor gene products (E-cadherin, PTEN, APC, and the Ink4 gene family members) and increased expression of transcription factors that promote the epithelial-to-mesenchymal transition. 34 CtBP is overexpressed in breast cancer, with high expression resulting in a lower median patient survival, an epithelial-to-mesenchymal transition, and lower genomic instability. 62 Currently, efforts are underway to treat cancer by suppressing CtBP activity. 63 An increased NADH/NAD ratio strengthens CtBP binding to its cellular targets, enhancing resistance to proteolytic digestion and promoting cell migration, partially through the metastases-promoting Tiam1 protein.64 -66 Increased iNampt expression increases intracellular NAD levels, while hypoxia increases the NADH/NAD+ ratio. Thus, increased tumor iNampt combined with hypoxia could lead to pro-carcinogenesis events via CtBP activation.13,64 -66 Van Horssen et al. 67 demonstrated that pharmacologic or genetic suppression of iNampt lowered NADH levels and glioma cell migration, while extracellular supplementation with NAD+ or iNampt re-expression abolished these effects. Cellular mobility was also associated with a lowered internal pH determined by the lactate dehydrogenase dependent pyruvate-lactate conversion, suggesting that tumor hypoxia may promote cell migration. Interestingly, iNampt is induced by hypoxia in breast cancer and hepatoma cells. 41 Thus, CtBP and iNampt might function cooperatively to promote carcinogenesis.
CD38
CD38 is an ADP ribosyl cyclase, first identified as a regulator of T-lymphocyte activation and proliferation. It is expressed at varying levels in B-cells, pancreatic acinar cells, smooth muscle cells, osteoclasts, and in different areas of the brain, eye, and gastrointestinal tract. Despite being an ectoenzyme, CD38 ablation in mice results in very high intracellular NAD+ levels, indicating that CD38 contributes to NAD+ homeostasis through constant degradation. 35 Higher CD38 expression is a negative prognostic marker in chronic lymphocytic leukemia (CLL), and anti-CD38 antibodies are in clinical trials to treat myeloma and CLL.68,69 Additionally, iNampt is overexpressed in myeloma, CLL, and other hematopoietic malignancies (but not in normal hematopoietic progenitor cells), which show high sensitivity to low concentrations of pharmacologic Nampt inhibitors.50,51,70 The role of CD38, NAD+, and Nampt in malignancy is presently poorly understood. However, it is likely that CD38 and Nampt function together in some malignancies, promoting the malignant phenotype.
Poly(ADP-ribose) polymerase-1 (PARP-1)
Poly(ADP-ribosyl)lation is a posttranslational protein modification that degrades NAD+ into NAM and ADP-ribose, forming long, branched ADP-ribose polymers at sites of broken DNA or at unusual DNA structures, such as cruciform DNA. This reaction is carried out predominantly by nuclear PARP-1, although there are at least 17 other PARP-1-related human proteins, 5 of which are also poly(ADP-ribose) polymerases and 12 that transfer single ADP-ribosyl units onto target proteins. PARP-1 has many, often-divergent cellular functions, including roles in regulating DNA repair, transcription, intracellular signaling, protein stability and degradation, as well as cellular proliferation, death, or differentiation. 36 PARP-1 activity protects cells from carcinogenesis, and the T2444C single nucleotide polymorphism, which reduces enzymatic activity by 30% to 40%, is associated with an increased incidence of prostate, lung, and esophageal cancers.36,71 -73 Massive DNA damage induces high PARP-1 activity rapidly degrading NAD+, resulting in cell death. 74 PARP-1 inhibition under these circumstances can prevent cell death.70,74 -77
PARP-1 activity is very high in malignant cells, with a roughly 45-fold higher activity than is seen in normal human lymphocytes, while the PARP-1 protein levels are roughly 23-fold higher. 75 This PARP-1 activity appears to be important in cancer cell survival and currently PARP-1 inhibition is being investigated as a possible cancer therapy (see below).78,79 In myeloma cells iNampt inhibition lowers cellular PARP-1 activity and cell viability, which is an event reversed by the addition of NAD+ precursors to the culture medium. 50 Additionally, high iNampt expression protects cells from death due to excessive NAD+ degradation caused by high tumor cell PARP-1 activity, an event partially mediated by SirT1 PARP-1 deacetylation and subsequent inactivation.10,50,74,80 -82 Thus, crosstalk between PARP-1 and iNampt plays a major role in cell viability during stress. 83 SirT1 also negatively regulates PARP-1 at the transcriptional level. 45 Since iNampt expression increases SirT1 activity, iNampt contributes to cell survival by attenuating PARP-1 activity. 11 Not surprisingly PARP-1 and iNampt overexpression often occurs in the same malignancy and is seen most of the malignancies listed in Table 1.58,84 -89 For the other malignancies PARP-1 expression either has not been examined or is not overexpressed. For some like myeloma iNampt inhibition significantly inhibits PARP-1 activity. 50 Last, in breast and prostate malignancies increased PARP-1 expression confers a worse prognosis.85,89
Elevated eNampt and Cancer
Plasma eNampt is elevated in a variety of human malignancies, including astrocytomas, myeloma, and male oral squamous cell; gastric, endometrial, hepatocellular, and colorectal carcinomas; and invasive breast cancer.47 -49,52,53,90 -93 Interestingly, plasma eNampt increases with increased astrocytoma grade and has been hypothesized to be a prognostic marker. 52 Similarly, eNampt is elevated at tumor higher stages in male oral squamous cell, hepatocellular, endometrial, and invasive breast carcinomas.47,53,93 Higher eNampt levels correlate with myometrial invasion and shorter patient survival in women with endometrial carcinoma. 44 Last, higher eNampt levels in invasive breast cancer correlated with lymph node metastases and the absence of estrogen and progesterone receptors. 93
The biological function of eNampt is presently poorly understood. Cardiac-specific eNampt overexpression in mice causes cardiac and cardiomyocyte hypertrophy by activation of the JNK1, p38, and ERK kinases. Interestingly, cardiomyocytes stressed in culture with H2O2 or serum starvation secrete eNampt. 25 Pretreatment of human chondrocytes with eNampt inhibited IGF-1 stimulated proteoglycan synthesis and AKT and insulin receptor substrate-1 phosphorylation, while activating ERK. 94 Zhang et al. 14 found that eNampt treatment rapidly induced interleukin-6 in murine macrophages, followed by interleukin-6-mediated Stat3 activation, an event that readily occurred even with mutated, enzymatically inactive eNampt. Interestingly, eNampt expression is induced in macrophages by interleukin-1β, tumor necrosis factor-α, and interleukin-6.95 -97 Constitutively activated Stat3 mediates dysregulated cell growth, survival, and angiogenesis, contributing to malignancy.98,99 Additionally, chronic inflammation, mediated by cytokines such as tumor necrosis factor-α and interleukin-6, with concomitant Stat3 activation, plays a prominent role in carcinogenesis, including many of the malignancies listed in Table 1.100,101 Taken together, these data suggests that plasma eNampt may contribute to carcinogenesis and tumor growth, partially explaining the increased eNampt accompanying human malignancies.47 -49,52,53,90 -93
Treatment of Human Malignancies With iNampt Inhibitors
The overexpression of Nampt in several human malignancies, combined with its promotion of many aspects of the malignant phenotype,37 -54 suggests that Nampt inhibition may exert anticancer effects. Hasmann and Schemainda 102 found that the highly specific, noncompetitive Nampt inhibitor FK866 induced delayed cell death by apoptosis in HepG2 human liver carcinoma cells with an IC50 of ~1 nM. The mechanism involved a gradual NAD+ depletion, which could be partially reversed by adding NAM or nicotinic acid. Others researchers found that Nampt inhibition also causes ATP depletion, lowered PARP-1 and SirT1 activities, and eventual cell death.102 -105 Interestingly, due to their increased NAD and ATP catabolism, tumor cells are more sensitive to iNampt inhibition than are benign cells. 106 Recently, several Nampt inhibitors have shown promise in treating several human malignancies and several are now in phase I and II clinical trials (Table 2).43,48,106 -110 Interestingly, Nampt inhibition by FK866 had little effect on cultured melanoma cells. 48 Several of these studies employed a Nampt inhibitor with another chemotherapeutic agent to induce “synthetic lethality,” where the inhibition of 2 gene products causes cell death, while inhibition of either gene product alone does not significantly lower cell viability. 111 For example, Bajrami et al. 78 employed an olaparib (a PARP-1 inhibitor) sensitization screen to examine the effect of inhibiting different enzymes involved in NAD metabolism and their effect on triple-negative breast cancer cell growth in murine xenografts. Nampt was identified as a nonredundant modifier of the olaparib response. Based on this the authors concluded that Nampt/PARP-1 inhibitor combinations may have value in treating triple-negative breast cancer.
Research Studies Demonstrating Nampt Inhibition May Have Some Clinical Efficacy in Treating Human Malignancies.

Conclusion
Both iNampt and eNampt are overexpressed in several human malignancies, where increased expression of either form is often associated with malignant progression.37 -54,90 -92 Additionally, several early studies indicate that Nampt inhibition may have clinical efficacy in treating some human malignancies.43,70,78,107,108 Based on these data, Nampt plays an important role in carcinogenesis and possibly cancer treatment, especially as it relates to other proteins involved in NAD metabolism. There are many aspects of Nampt biology that are presently poorly understood and need further study. For example, Santidrian et al. 112 found that a nonlethal reduction NAD+ levels by interfering with Nampt expression increased breast cancer metastases in an animal model, contradicting much of the data given above. Additionally, the same study revealed that mitochondrial complex I activity and the NAD+/NADH ratio regulated breast cancer progression. This last observation indicates that there are many aspects of NAD metabolism related to carcinogenesis and that Nampt activity in only one aspect of a complex NAD metabolome in cancer.
Footnotes
Acknowledgements
We would like to thank Jennifer Burton for help in preparation of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.