
Abstract
The p53 transcription factor regulates the expression of numerous genes whose products affect cell proliferation, senescence, cellular metabolism, apoptosis, and DNA repair. These p53-mediated effects can inhibit the growth of stressed or mutated cells and suppress tumorigenesis in the organism. However, the various growth-inhibitory properties of p53 must be kept in check in nondamaged cells in order to facilitate proper embryogenesis or the homeostatic maintenance of adult tissues. This requisite inhibition of p53 is performed primarily by the MDM oncoproteins, Mdm2 and MdmX. These p53-binding proteins limit p53 activity both in normal cells and in stressed cells seeking to promote resolution of their p53-stress response. Many mouse models bearing genetic alterations in Mdm2 or MdmX have been generated to explore the function and regulation of MDM-p53 signaling in development, in tissue homeostasis, in aging, and in cancer. These models not only have demonstrated a critical need for Mdm2 and MdmX in normal cell growth and in development but more recently have identified the MDM-p53 signaling axis as a key regulator of the cellular response to a wide variety of genetic or metabolic stresses. In this review, we discuss what has been learned from various studies of these Mdm2 and MdmX mouse models and highlight a few of the many important remaining questions.
Keywords
Introduction
The p53 transcription factor regulates the growth of mammalian cells by altering the expression of numerous genes whose products affect proliferation, senescence, cellular metabolism, and apoptosis.1,2 By reducing or eliminating the growth of cells bearing various forms of metabolic or genetic damage, these p53 effector pathways prevent the proliferation of mutated cells. Furthermore, p53 can participate in DNA repair in a transcription-dependent and -independent manner, possibly preventing or reducing the accumulated genetic defects in a stressed cell. 3 These various functions have led to the proposal that p53 acts as a tumor suppressor, in part, by “guarding the genome” to prevent the transmission of mutations to subsequent generations of cells. 4 Given that p53 affects so many different cellular functions directly or indirectly involved in cell growth, it is not surprising that mutation of the p53 gene is the most common genetic defect observed in human cancer.5,6
Since p53 activity limits the capacity of cells to divide, p53 activity must be kept under tight check both in nondamaged cells and in cells that have effectively repaired their damaged genome. Chief among the negative regulators of p53 are the MDM oncoproteins, Mdm2 and MdmX. These MDM proteins complex with the p53 transcription factor and inhibit p53-transactivation of heterologous p53-target genes. Interestingly, Mdm2 and MdmX are encoded by genes that are responsive to p53 transactivation.7-9 Thus, the level of p53 activity is autoregulated in cells due to the ability of p53 to induce the expression of Mdm2 and MdmX (Fig. 1). By inhibiting p53 activity, these MDM proteins promote the growth of normal cells, facilitating embryonic development and the maintenance of proper cell numbers in certain adult tissues. Furthermore, Mdm2, MdmX, and p53 form a signaling axis that can be differentially regulated by stress signals, allowing cells to alter their growth in response to various environmental cues.
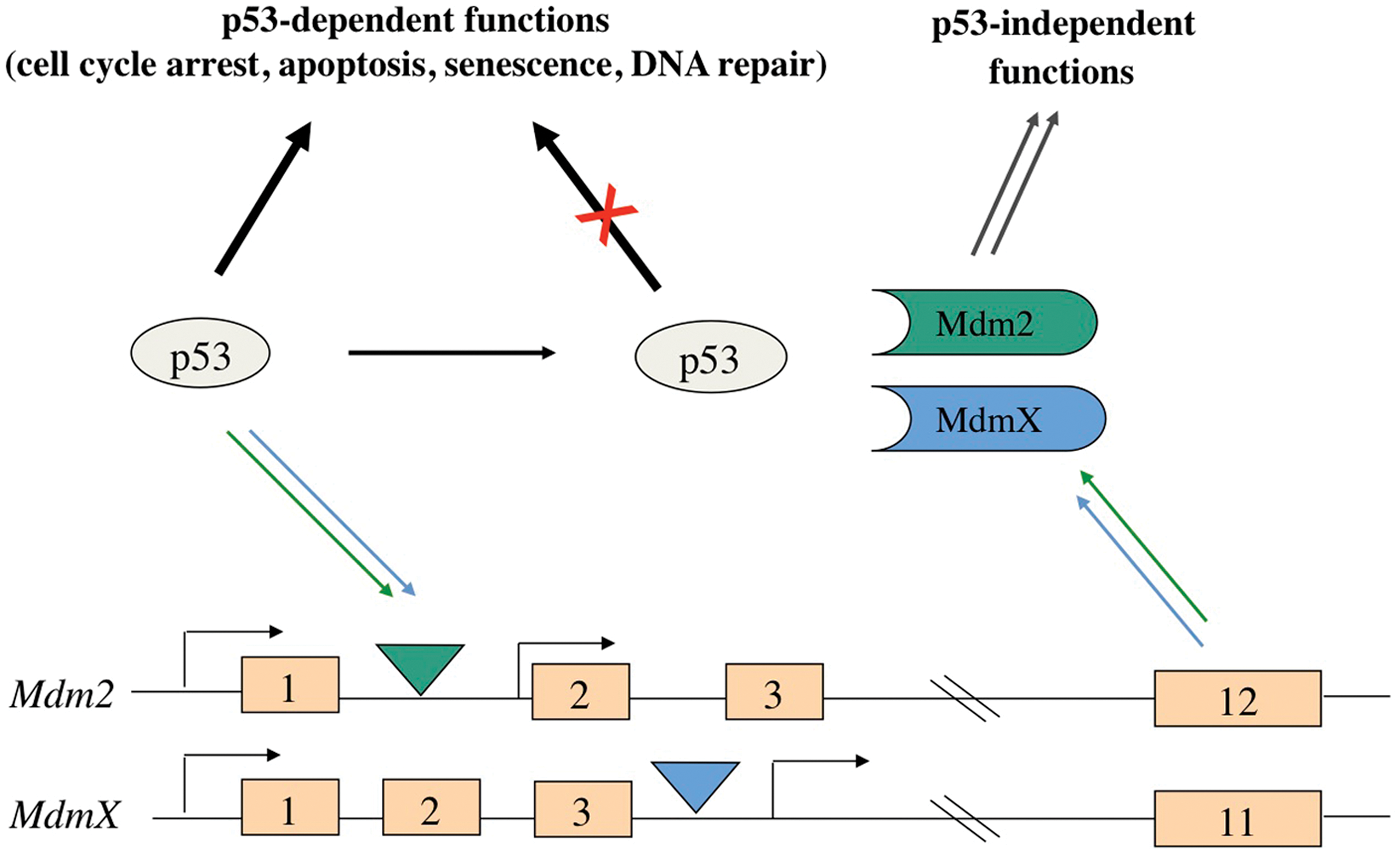
The Mdm2-MdmX-p53 signaling pathway. Mdm2 and MdmX expression is upregulated by p53 due to the presence of intragenic p53 promoter response elements (triangles). The subsequent increase in MDM proteins results in the inhibition of p53 functions due to p53 and Mdm2/MdmX complex formation.
The importance of the MDM signaling pathway in regulating normal cell growth is underscored by the high frequency in which p53 mutations or anomalous levels of MDM2 or MDMX are observed in human cancer. For example, somatic missense mutations in the p53 gene have been detected in a very broad spectrum of human tumors, and Li-Fraumeni syndrome patients bearing germline p53 mutations display greatly increased genomic instability and a vast predisposition to developing cancer.10,11 In contrast, MDM2 and MDMX genes are rarely lost or mutated in human tumors. Rather, these MDM genes are amplified and/or overexpressed in a significant percentage and wide variety of human cancers. Overexpression of these p53 inhibitors results in a functional loss of p53 activity in the tumor cells. In addition, p53-independent roles for MDM proteins in neoplasia have been described recently, suggesting that members of this signaling axis may have an even greater impact on disease than previously expected.
Over the past 15 years, many mouse models bearing genetic alterations in Mdm2 or MdmX have been generated to probe the role of the Mdm2-p53 signaling in development, in tissue homeostasis, in aging, and in cancer. Analysis of these individual mouse models and those generated by the intercrossing of several models has provided either convincing genetic support for preceding in vitro analysis of MDM protein functions or direct evidence refuting the conclusions drawn from in vitro studies. Furthermore, analysis of mouse models has revealed novel physiological roles for this well-established signaling pathway. Here we review the various Mdm2 and MdmX mouse models that have been generated to explore MDM regulation of p53, and we highlight some of the critical insights provided by these studies that have expanded our understanding of MDM-p53 regulation. In addition, we present several mouse studies that have indicated potential p53 independent roles for MDM proteins. To better discuss the role of the MDM-p53 signaling axis in normal and abnormal cell growth, we begin with a brief review of p53-null mice.
The Basics: p53 Knockout Mice
Early in vitro studies suggested that p53 was a critical regulator of the cell cycle, and p53 knockout mice were anticipated to have severe defects in embryonic development and cellular growth.12-15 However, p53-null mice proved to be surprisingly viable and physically indistinguishable from wild-type littermates at an early age. 16 Importantly, these mice rapidly developed spontaneous tumors, confirming a critical role for p53 in tumor suppression. These tumors were mostly lymphomas (chiefly T cell in origin), with some incidence of sarcomas and other tumor types. Subsequent analysis of primary fibroblasts derived from this model revealed that p53 reduction or deletion could upregulate cell proliferation and inhibit the growth arrest of cells after DNA damage, revealing a role for p53 in the DNA damage response. 17 Other groups developed additional p53 knockout mouse models that also formed spontaneous tumors with a relatively similar spectrum.18,19 These subsequent studies observed that a subset of p53- null mice die in utero due to exencephaly and that thymocyte apoptosis normally induced by exposure to ionizing radiation was greatly compromised by p53 deficiency, underscoring a critical role for p53 in regulating cell death in response to DNA damage.19,20 Interestingly, the tumorigenic phenotype of p53+/– heterozygous mice differed from p53-null mice, with heterozygous mice displaying neoplasia at a later age and forming mostly sarcomas, with a lessor percentage of lymphomas and other tumor types. As anticipated from earlier studies of familial cancer syndromes, loss of heterozygosity (LOH) for the single wild-type p53 allele could be detected in tumors isolated from p53 + /– mice. 18 However, subsequent work determined that only half of the tumors isolated from p53-heterozygous mice undergo LOH, suggesting that perturbation of other members of the p53 signaling axis might account for functional loss of p53 in the tumors or that a simple reduction in p53 function is sufficient to increase the susceptibility of the cell to tumorigenesis. 21 Collectively, these initial studies of p53-null and p53-heterozygous mice demonstrated the importance of p53 in governing the DNA damage response and in suppressing tumor formation, and they validated the use of mouse modeling as a powerful tool to study both tumor suppressor genes and the p53 signaling pathway in vivo.
Numerous laboratories have continued to use p53-deficient and p53-knockin mouse models to successfully uncover multiple roles for p53 in the regulation of cell growth and tumorigenesis. Although beyond the scope of this review, these p53 models have greatly assisted in our understanding of the tumorigenic effects of p53 loss of function and gain of function mutations and have highlighted roles for p53 in other biological settings, including reproduction, development, and aging. Many of these in vivo p53 functions are discussed in further detail in several excellent and recent reviews of p53 mouse model phenotypes.22-25
Mdm2 Knockout Mice
The Mdm2 oncoprotein was the first cellular negative regulator of p53 discovered and studied in depth.26-28 Mdm2 was identified and cloned as one of several genes present on a double minute chromosome that was isolated from a spontaneously transformed 3T3 cell line. 29 Subsequent transfection studies revealed that Mdm2 protein could bind to the p53 transcription factor and sequester it away from p53 target gene promoters in vitro.26-28 Interest in the oncogenic potential of Mdm2 was greatly increased when the Mdm2 gene was found amplified in a significant fraction of human sarcomas and in a number of human tumors of the lung, breast, bone, and soft tissues.30,31 It is thought that Mdm2 constitutively inhibits wild-type p53 in these tumors, as elevated Mdm2 expression and concurrent p53 mutations are rarely observed.
To study the role of Mdm2 in vivo, 2 separate Mdm2 knockout mouse models were generated.32,33 Unlike p53-null mice, which undergo relatively normal development, Mdm2-null mice die post-implantation at E5.5-6.5 of early development. The embryos appeared much smaller than wild-type littermates and displayed a highly disorganized architecture. These developmental defects were due to unregulated p53 activity in the embryo, as concomitant knockout of p53 completely rescued the early developmental lethality of Mdm2-null mice.32,33 These results clearly documented a critical role for Mdm2- dependent regulation of p53 during development. Although it remains to be formally demonstrated, increased apoptosis in cultured E3.5 blastocysts harvested from intercrosses of Mdm2 + /– mice suggests that loss of Mdm2 function results in the embryonic demise of post-implantation embryos due to unregulated p53-dependent apoptosis. 34 In keeping with this possibility, our laboratory has recently determined that deletion of certain growth inhibitory genes such as Rb or the p53-responsive Cdkn1a (p21) gene fails to rescue Mdm2-null embryos beyond E6.5 gastrulation. Mice deleted for both Mdm2 and p53 are viable, and they form spontaneous tumors with a similar tissue spectrum as p53-null mice. 35 Since the phenotype of Mdm2/p53 double-null mice and primary cells is indistinguishable from p53-null mice, it would appear that the chief role of Mdm2 in cell growth is to act as a negative regulator of p53 activity. 35
Transfection analysis of Mdm2 and p53 functions using human cells and mouse fibroblasts generated from Mdm2/p53 double-null embryos subsequently determined that Mdm2-p53 binding promoted the destabilization of p53, providing a second potential mechanism for Mdm2-mediated inhibition of p53 function.36,37 Subsequent in vitro work determined that the Mdm2 protein contains a carboxy-terminal RING domain with E3-ubiquitin ligase activity.36-39 Furthermore, Mdm2 was found to add ubiquitin moieties to p53 and induce p53 nuclear export degradation in the 26S proteasome. 38 Consistent with these results, p53 protein levels are often elevated in tissues of mice partially or fully ablated for Mdm2 (as discussed below).
Mdm2-Hypomorphic Mice
In addition to these early studies of Mdm2-null mice, an Mdm2-hypomorphic allele (Mdm2 puro ) was generated that provided valuable insight into the role of Mdm2 regulation of p53 in adult tissues. 40 A majority of heterozygous mice bearing an Mdm2-hypomorphic allele and an Mdm2-null allele (Mdm2 puro/ ≠ 7-9 ) were viable, indicating that the Mdm2 hypomorph protein partially retained wild-type Mdm2-dependent regulation of p53. However, these Mdm2-compromised adult mice had decreased body weights and defects in hematopoiesis. In agreement with a phenotype that was p53-dependent, the mice also displayed increased endogenous p53 target gene expression and spontaneous apoptosis. Furthermore, Mdm2 puro/ ≠ 7-9 mice exhibited increased sensitivity to p53-dependent responses resulting in greatly decreased survival after whole-body irradiation. This in- creased IR-induced lethality was shown to be solely dependent on p53, as Mdm2 puro/ ≠ 7-9 , p53-null mice were completely radio-resistant.
MdmX-Knockout Mice
MdmX, a close protein family member of Mdm2, was also shown to be a critical regulator of p53 in vivo. MdmX and Mdm2 share 34% protein homology, with each containing homologous p53- binding, acidic, zinc finger, and RING finger domains (Fig. 2). 41 Like MDM2, the MDMX gene is amplified and/or overexpressed in a number of different human tumor types, including breast cancer, brain and soft tissue tumors, and retinoblastoma.42-46 However, in vitro studies have determined that MDMX does not mediate p53 protein degradation, suggesting that MDMX inhibits p53 activity solely through steric inhibition of p53 transcriptional activity.47,48
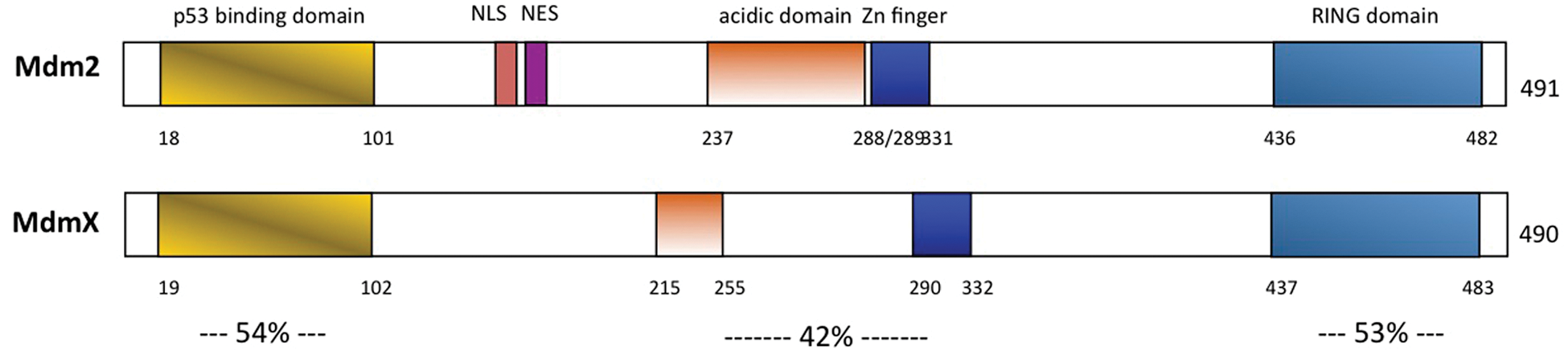
Proposed functional domains of the Mdm2 and MdmX oncoproteins. The percentages of identities of the individual domains shared by the 2 MDM proteins are shown. NLS = nuclear localization signal; NES = nuclear export signal; Zn finger = Zinc finger.
Several groups have generated MdmX-null alleles in mice.49-51 In the initial study, MdmX-null mice displayed an embryonic lethality at E8.5-9.5. 49 A subsequent and different MdmX-null model displayed a slightly later time of lethality: E10.5-11.5 of gestation. 50 As with the Mdm2-null models, these MdmX-null mouse models can be rescued by co-deletion of p53. Differences in the time of lethality between the 2 MdmX-null models may be due to subtle differences in the genetic backgrounds of mice in these studies or may arise due to slight differences in the MdmX-targeted alleles themselves. In keeping with this latter possibility, Mdm2/MdmX compound heterozygous mice (Mdm2 + /– , MdmX + /– ) have been reported to have an embryonic or neonatal lethal phenotype, although we and other groups using different MdmX-null alleles can readily generate and breed the Mdm2/MdmX compound heterozygous mice. 52 In addition, there are differences in the timing of MdmX-null embryonic demise (E7.5 versus E14-15) when Cdkn1a (p21) is co-deleted (see below).34,53 It remains to be determined why these subtle differences exist between the phenotypes of the various MDM models.
The difference in the time of lethality between Mdm2-null mice (E5.5-6) and MdmX-null mice (E8.5-11) indicates differences in the requirements for Mdm2- and MdmX-mediated inhibition of p53 during development. Furthermore, MdmX-null lethality in mice appears to be caused by a lack of cellular proliferation rather than by an increase in apoptosis (as seen in Mdm2-null embryos), and subsequent work has revealed that co-deletion of Cdkn1a can delay the embryonic lethality of MdmX-null mice to E13.5-15.5. 54
Other differences are also apparent between the phenotypes of Mdm2-null and MdmX-null mice. Although deletion of p53 rescued either MDM-null model from embryonic lethality and yielded mice that developed a spectrum of spontaneous, p53-null like tumors, MdmX/p53 double-null (but not Mdm2/p53 double-null) mice displayed a faster rate of tumorigenesis than p53-null mice.35,53 These results uncover a p53-independent, tumor-suppressing role for endogenous levels of MdmX that exists in addition to the oncogenic ability of MdmX to inhibit p53 activity. 53 In support of this possibility, mouse embryonic fibroblasts (MEFs) derived from mouse models co-deleted for MdmX and either p53 or p21 proliferate faster than p53-null MEFs or p21-null MEFs and also display increased chromosomal instability.53,54 More work is needed to elucidate the precise nature of this p53-independent role for MdmX.
Advanced Models: Mdm2 and MdmX Conditional Mice
Although the results from the Mdm2 and MdmX knockout mouse studies demonstrated the critical roles for each in regulating p53 during gestation, the embryonic lethal phenotypes of these models preclude further analysis of MDM functions in latter stage of development or in adult tissues. To further explore the roles of Mdm2 and MdmX in vivo, Mdm2 and MdmX conditional knockout mouse models have been generated.55-57 These models allow temporal and/or spatial-restricted ablation of MDM proteins in mice, circumventing the early embryonic lethality in the constitutive MDM-null models. Using these loxP-based conditional models and various transgenic Cre-driver mice, Mdm2 and MdmX expression has been subsequently ablated in many different tissue types. A summary of the results of these published studies is provided in Table 1.
Conditional Ablation of Mdm2 and/or MdmX in Mice
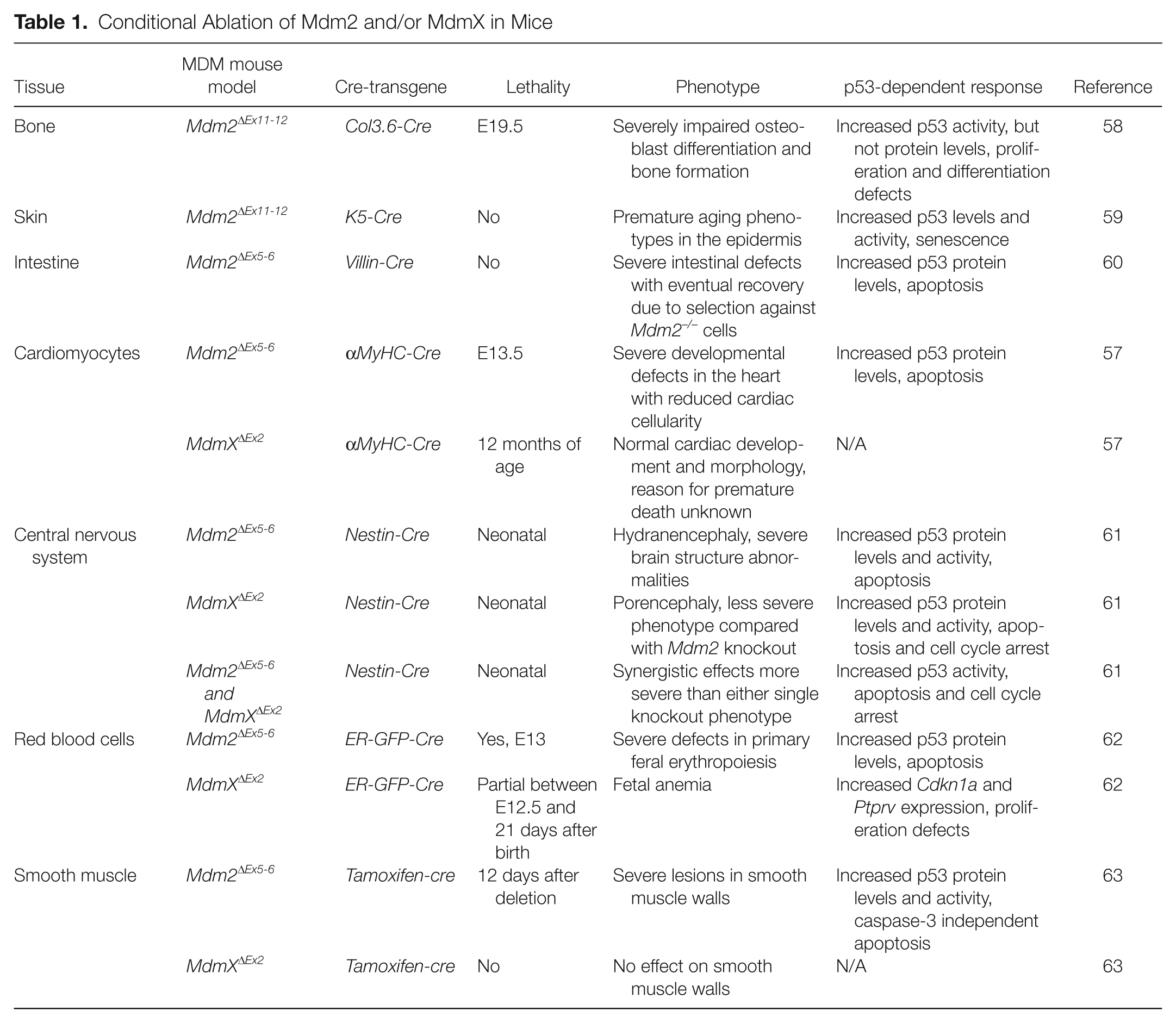
Analysis of these models has revealed major differences in the requirements of MDM proteins in the development of different tissue types. For example, both Mdm2 and MdmX are critical for development of the central nervous system in mice. 61 Furthermore, absence of both Mdm2 and MdmX resulted in a more severe phenotype than either single knockout, suggesting synergistic effects in this tissue. Conversely, deletion of Mdm2 in cardiomyocytes led to embryonic lethality, whereas deletion of MdmX in these cells resulted in no obvious developmental defects. 57 These models also suggest that Mdm2 and MdmX inhibit different p53-dependent responses in a tissue-specific manner. For example, whereas Mdm2 clearly regulates p53-dependent apoptosis in adult intestine, cardiomyocytes, red blood cells, and embryonic epithelium, deletion of Mdm2 in osteoblast progenitor cells results in upregulated Cdkn1a expression and a p53-dependent cell growth arrest, with no increase in apoptosis.34,57,58,60,62 In contrast, deletion of Mdm2 in epithelial cells of adult mice induces neither a transient proliferative arrest nor apoptosis but promotes p53-dependent cell senescence in the epithelial stem cell compartment. 59 Thus, the mechanism by which Mdm2 (or MdmX) regulates cell growth in a given tissue, whether by promoting cell proliferation, inhibiting apoptosis, or preventing premature senescence, is highly dependent upon the precise role of p53 in the biology of that specific tissue.
Mdm2 and MdmX Knockin Mice: Exploring RING Domain Function
Mouse models have been used to study the roles of specific Mdm2 and MdmX protein domains. These models were developed as valuable tools not only to confirm or refute the physiological relevance of the initial experimental results in vitro but also to determine whether specific domains should be favored as therapeutic targets to effectively manipulate p53 signaling. As stated above, Mdm2 negatively regulates p53 via 2 mechanisms: direct protein binding and E3-ligase-dependent ubiquitination. To differentiate the importance of these 2 distinct functions, a mouse model with a specific knockin mutation in the RING domain of Mdm2 (C462A) was generated. 64 Co-immunoprecipitation experiments indicate that this mutation specifically abolished the E3-ligase activity of Mdm2 without altering the p53 binding function. Surprisingly, these mice die very early in embryonic development at E7.5 in a p53-dependent manner. Although this proved problematic for further experimentation, the lethality in these mice demonstrated that binding of Mdm2-p53 is not sufficient to inhibit p53 activity during development. It remains to be seen whether Mdm2 E3 ligase activity is required for Mdm2-inhibition of p53 activity at later times in development or in adult mice. Although most of the phenotypes described in the various Mdm2-conditional studies listed in Table 1 can be attributed to excess p53 activity in the cell, not all tissues ablated for Mdm2 in these studies displayed increased p53 protein levels, and it is possible that a ligase-dead Mdm2 might still functionally inhibit p53 in certain tissues or in adult mice. 58
Although MdmX and Mdm2 both contain a carboxy-terminal RING domain, MdmX does not appear to share the intrinsic E3 ligase activity that Mdm2 possesses for p53. Rather, MdmX has been proposed to block p53 activity by binding and inhibiting the transactivation domain of p53 and/or by forming a heterodimer with Mdm2 and promoting p53-ubiquitination indirectly by enhancing the E3 ligase activity of Mdm2.41,48,65,66 Two mouse models with MdmX RING domain mutations have been generated to further explore the function of this protein domain. One model is a conditional MdmX RING domain knockout in which the endogenous MdmX exon 11 was replaced with a modified exon 11 that is missing sequences encoding 49 amino acids from the RING domain. 67 Mice homozygous for the RING deletion allele (MdmX ≠ RING/ ≠ RING ) did not develop past E9.5 due to aberrant p53 activation, so these mice were crossed to a conditional hypomorphic p53 model that restored wild-type p53 levels in the presence of Cre-recombinase. MEFs generated from these mice showed that the MdmX RING domain was dispensable for the ubiquitination and degradation of Mdm2 and p53, suggesting that the lethality in MdmX ≠ RING/ ≠ RING mice may be due to a lack of p53 transcriptional inhibition. It is interesting to note that ablation of the MdmX RING domain in adult tissues had no noticeable impact on tissue function, suggesting a singular role for the MdmX RING domain in embryonic development.
Interestingly, data from a concurrent study, which substituted a single amino acid in the endogenous murine MdmX RING domain (C462A), confirmed some results from the MdmXΔRING/ΔRING model while refuting others. 68 Mouse embryos that were homozygous for the RING domain mutation (MdmX C462A/C462A ) were not recovered past E9.5 due to p53-dependent apoptosis and cell proliferation arrest, exactly replicating the embryonic lethality of the MdmX ≠ RING/ ≠ RING model. Likewise, the MdmX C462A/C462A embryos displayed increased p53 target gene expression that likely contributed to the embryonic lethal phenotype. However, analysis of Mdm2 in MEFs generated from MdmX C462A/C462A , p53-null mice indicated that an intact MdmX RING domain was required for full MdmX-Mdm2 binding and Mdm2 ubiquitination, contrary to what was seen in MdmX ≠ RING/ ≠ RING MEFs. Although it is clear that the MdmX RING domain is critical in limiting p53 activity during development, the precise role of the MdmX RING in p53 inhibition is still open to debate. New MdmX models may be required, including those mutated for p53 recognition but retaining the RING domain. Such mice might be useful in resolving the precise contribution of the MDMX RING to the inhibition of p53 function in human tumors.
Mdm2 and MdmX Knockin Mice: Exploring Cellular Stress Responses
The above studies clearly illustrate the important roles of MDM proteins in inhibiting p53 activity during development and in normal tissue growth. However, in order for p53 to become fully activated in response to various forms of cellular stress, MDM inhibition of p53 must be temporarily attenuated to facilitate p53 protein stabilization and subsequent target gene activation. This rapid, transient activation of p53 was long thought to rely on post-translational modifications of numerous proteins within the p53 pathway. These modifications might be induced by various kinases or acetyl transferases activated in a stress-specific manner and would facilitate the rapid and specific response of cells to different types of stress.
Activation of the p53 transcription factor during the DNA damage response has been the focus of numerous in vitro and in vivo studies, and many molecular models integrating DNA damage signaling with p53 stabilization and activation have been proposed. Numerous DNA damage responsive proteins recognize DNA breaks, caused by ionizing radiation (IR) or cytotoxic drugs, and mediate downstream pathways such as DNA repair and apoptosis. The ataxia telangiectasia mutated (ATM) kinase is a major effector of the DNA damage response that phosphorylates a wide variety of protein targets. It has been previously shown that p53 protein stabilization is dependent on functional ATM signaling, and p53 encodes several residues in or adjacent to the Mdm2-interaction domain that are known to be direct or indirect targets of ATM phosphorylation. Several p53-knockin mouse models have revealed that ATM phosphorylation of p53 is necessary for the full transcriptional activity of p53 after DNA damage, particularly those genes involved in apoptosis. However, these modifications had only modest effects on p53 protein stability and p53-mediated tumor suppression.25,69-71
Because activated ATM also phosphorylates Mdm2 and MdmX, it was proposed that ATM modification of MDM proteins might potentially regulate IR-induced p53 stabilization. ATM was shown to phosphorylate MDM2 serine residue 395 in vitro, and an antibody that binds MDM2 only when MDM2-S395 is unphosphorylated confirmed that MDM2 phosphorylation occurs in human cells after DNA damage. 72 To examine the physiological effects of this post-translational modification on p53 regulation, 2 knockin Mdm2 mouse models have been generated recently. 73 One of these mutant Mdm2 alleles contains a serine to alanine substitution at murine serine residue 394 (homologous to human serine 395). Mice homozygous for the mutation (Mdm2 SA/SA ) were viable, with no differences observed in the development, growth, or fecundity of untreated animals. However, p53-dependent DNA damage responses were greatly reduced in these mice, as evidenced by a lack of p53 protein stabilization, p53 target gene induction, and p53 apoptotic and cell cycle arrest function. Furthermore, the lack of p53 function in Mdm2 SA/SA mice resulted in complete radio-resistance and increased spontaneous tumorigenesis, revealing the importance of this Mdm2 phosphorylation event in p53 regulation. An additional Mdm2-S394 model was generated to mimic constitutive phosphorylation by substituting this ATM-target serine with an aspartic acid residue. These Mdm2 SD/SD mice were viable and indistinguishable from controls, indicating that this single modification did not affect Mdm2-p53 regulation in the absence of DNA damage. Interestingly, whereas the early DNA damage response was unaltered in these mice and p53 suppression of spontaneous tumorigenesis was restored, p53 protein stability and activity were prolonged in Mdm2 SD/SD mice, indicating that Mdm2 serine 394 phosphorylation controls the duration of the p53 response in vivo.
MdmX was also proposed to be a target for DNA damage-induced phosphorylation by multiple kinases, including ATM (serine residue 402 in mice) and Chk2 (serine residues 341 and 367 in mice). All 3 of these serine residues were mutated to alanine in a triple knockin mouse model (MdmX 3SA ) to study the effect of DNA damage-induced phosphorylation of MdmX on p53 function. 74 Similar to what was observed in the Mdm2 SA/SA model, MdmX 3SA/3SA mice had impaired p53 stabilization and decreased p53 activity in response to IR. In addition, these MdmX knockin mice displayed impaired MdmX degradation. Most MdmX 3SA/3SA mice were radio-resistant to normally lethal doses of IR, and these phosphorylation events were necessary for robust suppression of lymphomas induced by an Eμ-Myc transgene. However, in contrast to the Mdm2-S394A knockin model, spontaneous tumorigenesis was not observed in MdmX 3SA/3SA knockin mice. Differences in the ability of Mdm2 and MdmX to regulate p53 activities following DNA damage are consistent with differences in the phenotypes of Mdm2-null and MdmX-null mice: MDM proteins clearly have different roles and capacities for regulating p53. However, the contributions of other Mdm2 and MdmX post-translational modifications to p53 regulation and p53 tumor suppression following DNA damage remain to be explored.
Abnormal ribosomal biogenesis is another cellular stress very recently shown to activate p53. It has been known for some time that the Mdm2 protein can interact in vitro with a variety of ribosomal proteins such as L5, L11, and L23, and structure-function studies determined that these interactions occur at the Mdm2 zinc finger (ZF) domain.75-80 To study the physiological significance of these protein-protein interactions, the ZF domain was disrupted (Mdm2 C305F ) in mice. 81 Since these mice are viable and have a normal lifespan, the Mdm2 C305F/C305F knockin mice appear to retain wild-type Mdm2 function during development, and homeostatic and DNA damage-induced p53 functions were unchanged in adult Mdm2 C305F/C305F mice. However, p53-dependent cell cycle arrest in response to ribosomal stress induced by the transcriptional inhibitor actinomycin D was greatly perturbed in Mdm2 C305F/C305F mouse embryonic fibroblasts. In vivo co-immunoprecipitation experiments confirmed that Mdm2-C305F protein does not bind to L5 or L11, although it can still bind with L23. Additionally, tumorigenesis was significantly increased in Mdm2 C305F/C305F mice crossed with mice bearing the Eμ-Myc transgene, confirming Mdm2 as a major mediator of the ribosomal stress response in vivo.
Addressing stress-induced modifications of Mdm2 and MdmX on MDM-p53 signaling in mice is a relatively new area of scientific exploration in the MDM field. Based upon the rather large phenotypic impact in mice of the few MDM modifications modeled to date, it would appear that the MDM-p53 signaling axis truly serves as a critical node for the conversion of stress signals into alterations in p53 functions. Clearly, additional in vivo explorations of the effects of post-translational modifications to Mdm2 and MdmX are warranted.
Mdm2 and MdmX Transgenic Mice
Mice bearing increased levels of Mdm2 or MdmX expression have been generated to further explore MDM-p53 signaling and to model MDM gene amplification events frequently observed in human tumors. These models have been surprisingly difficult to generate, especially given that p53-null mice are viable. This difficulty may be partly due to the unusual toxicity frequently seen in primary cells induced to overexpress these proteins. 82 However, several overexpressing MDM models have been successfully produced and studied. The first was an Mdm2-overexpressing mouse model that was generated by introducing a cosmid containing the mouse Mdm2 gene (including transcriptional start sites, introns, and 3′ sequences) into mouse embryonic stem (ES) cells. 82 ES cell clones were selected that had multiple copies of Mdm2 integrated into a single site in the genome. These cells were used to generate chimeric mice, which were then bred to generate a line of Mdm2-transgenic mice that have a 3- to 5-fold increase in Mdm2 expression levels in various tissues. Interestingly, ES cell lines generating higher levels of Mdm2 expression proved to be incapable of even generating chimeric mice, suggesting that large increases in Mdm2 are incompatible with normal embryonic cell biology.
As was seen in human cancers, overexpression of Mdm2 in mice led to spontaneous tumorigenesis. 83 This result demonstrated that overexpression of Mdm2 was a major contributor to tumorigenesis and not just a “passenger” event in human cancers. Furthermore, the mice presented with a broad spectrum of spontaneous tumors similar to those seen in p53-null mice, and p53 activity and protein levels were diminished in tissues and cells derived from these mice. Sequence analysis confirmed that the Mdm2 transgene was wild-type, revealing that Mdm2 did not require additional “activating” mutations to promote tumorigenesis when overexpressed. Surprisingly, Mdm2-transgenic, p53-null mice displayed a somewhat different tumor spectrum than p53-null mice. These results indicate the existence of a p53-independent role for Mdm2 in tumorigenesis when overexpressed, at least in cells with inactivated p53. Additional evidence of a p53-independent role for Mdm2 in tumorigenesis was seen in studies of tumor-derived Mdm2 splice variants. These variants were found to promote tumorigenesis in mice, even though these splice-isoforms encoded Mdm2 proteins that lack the p53-binding domain.84,85 More recent studies using Mdm2 transgenic mice have revealed a role for Mdm2 in DNA break repair and chromosomal damage in cells lacking p53.86,87 The relative contributions of p53-dependent signaling and p53-independent mechanisms in the neoplastic transformation of cells are important topics for future studies.
Subsequent crosses of Mdm2-transgenic mice with MdmX + /– mice determined that overexpression of Mdm2 can fully rescue the embryonic lethality of the MdmX-null model. 88 As it was previously established that deletion of p53 could rescue the embryonic lethality of MdmX-null mice, the ability of the Mdm2 transgene to inhibit p53 activity in mice likely underlies the recovery of the Mdm2-transgenic, MdmX-null mice.49,50 These results further reveal that MdmX is not absolutely required for Mdm2 to properly inhibit p53 function. In keeping with this conclusion, Mdm2-transgenic, MdmX –/– fibroblasts lack detectable levels of p53 protein. 88
Efforts to understand the role of MdmX amplification in tumorigenesis have produced mixed results. One study generated a conditional MdmX transgene (MdmX Tg ) driven by the chicken β-actin promoter and CMV immediate-early enhancer. 89 This conditional allele allowed temporal control over widespread MdmX overexpression to avoid the problems encountered in the Mdm2-transgenic model. However, it was determined that MdmX overexpression in this model did not lead to embryonic defects, and a subsequent constitutive MdmX-transgenic model (MdmX Tg15 ) was generated. Both the MdmX Tg and MdmX Tg15 mice displayed increased tumorigenesis with a predominance of sarcomas, verifying that MdmX is an oncogene in vivo.
Interestingly, the results of a separate ROSA26-driven, Myc-tagged MdmX transgenic model (MdmX T ) revealed a very different phenotype. 90 Inheritance of one MdmX transgene (MdmX T/+ ) resulted in elevated MdmX levels in the hemizygous mice but did not increase spontaneous, DNA damage-induced, or Eμ-Myc-induced tumorigenesis. This may be due to MdmX levels remaining below a certain threshold to affect tumorigenesis. However, mice homozygous for the transgene (MdmX T/T ) die early in embryonic development and present with severe vascular defects. Unlike the typical lethality seen in other mouse models with varying Mdm2 and/or MdmX levels, the lethality in the MdmX T/T mice was completely p53-independent. This may be due to the high levels of MdmX or to aspects of the transgene unrelated to MdmX itself. A side-by-side comparison of the MdmX levels in the 2 MdmX-transgenic models might explain the vast differences in the phenotypes observed in these 2 studies.
Mouse Models Exploring Mdm2 Transcriptional Regulation
Recently, human populations with a specific single nucleotide polymorphism (SNP) in the intragenic MDM2 promoter were found to have an increased incidence of cancer. 91 The SNP309G polymorphism resulted in an increased level of MDM2 expression compared with levels with the SNP309T polymorphism. The presence of these base pair alterations in the MDM2 promoter region provides a potential alternate mechanism for MDM2 upregulation and subsequent tumor induction, a mechanism that is distinct from an increase in MDM2 gene copy number. To test the role of this SNP in tumor incidence, mouse models bearing knockin alterations in the second intron of Mdm2 were generated. 92 These mice retained wild-type Mdm2 coding sequences but possessed different human variants of the MDM2 promoter (harboring either the SNP309T or SNP309G polymorphism). Mice homozygous for the SNP309G (Mdm2 SNP309G/G ) had high Mdm2 expression levels and decreased p53 function in multiple cell types compared with the wild-type and Mdm2 SNP309T/T mice. Importantly, the Mdm2 SNP309G/G mice had significantly higher rates of tumorigenesis compared with the Mdm2 SNP309T/T model, corroborating the increased incidence of human cancer patients who contain this promoter variant.
Conclusions
Much has been learned regarding the in vivo role of MDM-p53 signaling in development, in tissue homeostasis, and in tumorigenesis. Work in mice has demonstrated the importance of Mdm2 and MdmX in regulating p53 activity in normal cells and how perturbation of this pathway can readily lead to cancer. Although numerous other proteins have the potential (and, in some cases, have been directly shown) to also govern p53 activity in cells, the results of these Mdm2 and MdmX genetic studies have clearly identified the MDM proteins as the chief negative regulators of p53 in development. Furthermore, recent work with various Mdm2 and MdmX-knockin models has just begun to highlight the critical role of MDM post-translational modification in regulating the response of cells to DNA damage or other forms of stress.
Many important questions regarding MDM-p53 signaling remain unanswered, including the role of MDM-p53 signaling in cell metabolism, the precise role and requirement of MdmX in proper MDM-p53 signaling, the nature and significance of Mdm2 splice variants in cancer, and the impact of the p53-independent effects of Mdm2 and MdmX on cell transformation. Fortunately, the MDM scientific community remains a collegial group that is always ready to share results and resources, thus facilitating the tackling of important questions using mouse models. For this reason, the mice described in this review or primary cells generated from these models have proven to be widely shared and highly useful tools for many different laboratories aiming to explore MDM-p53 signaling. In addition, these models stand as a testament to the power and utility of genetic engineering in mice to study molecular signaling pathways perturbed in human cancer.
Footnotes
Acknowledgements
We thank Zdenka Matijasevic and Jean-Christophe Marine for helpful discussion.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This publication was supported by a grant from the NIH/NCI to SNJ (RO1-CA077735).