
Abstract
The p53 tumor suppressor is highly responsive to different physiological stresses such as abnormal cell proliferation, nutrient deprivation, and DNA damage. Distinct signaling mechanisms have evolved to activate p53, which in turn modulate numerous pathways to enhance fitness and survival of the organism. Elucidating the molecular mechanisms of these signaling events is critical for understanding tumor suppression by p53 and development of novel therapeutics. Studies in the past decade have established that MDM2 and MDMX are important targets of signaling input from different pathways. Here, we focus our discussion on MDM2 and MDMX phosphorylation, which is important for p53 activation by DNA damage. Investigations in this area have generated new insight into the inner workings of MDM2 and MDMX and underscore the importance of allosteric communication between different domains in achieving an efficient response to phosphorylation. It is likely that MDM2 and MDMX regulation by phosphorylation will share mechanistic similarities to other signaling hub molecules. Phosphorylation-independent p53 activators such as ARF and ribosomal proteins ultimately achieve the same outcome as phosphorylation, suggesting that they may induce similar changes in the structure and function of MDM2 and MDMX through protein-protein interactions.
MDM2 and MDMX Are Key Regulators of p53
A unique feature of the p53 tumor suppressor is its stabilization after exposure to many stress signals. This leads to induction of numerous transcriptional targets that inhibit cell cycle progression, induce apoptosis, and regulate energy metabolism. 1 The MDM2 and MDMX proteins are possibly responsible for establishing most of the dynamic features of the p53 pathway. It can be argued that the ability of p53 to act as a major tumor suppressor is in part due to its regulation by MDM2 and MDMX. MDM2 is best known as an ubiquitin E3 ligase for p53 that promotes p53 degradation.2,3 Although additional E3 ligases (Pirh2, Cop1) have also been shown to degrade p53 in cell culture,4,5 mouse models provided strong evidence that MDM2 function is indispensable for controlling p53 activity at all stages of life.6-8 The role of MDM2 as a major regulator of p53 stability is also validated by small molecule inhibitors that disrupt p53-MDM2 binding.9,10
The MDM2 homolog MDMX is also emerging as an important regulator of p53. 11 The physiological significance of MDMX was revealed by the embryonic lethality of MDMX-null mice due to activation of p53.12-14 Tissue-specific knockout of MDMX generally results in mild phenotypes compared to MDM2,7,15,16 suggesting a supplemental function in p53 regulation. In fact, MDMX deficiency can be compensated by transgenic expression of additional MDM2 proteins in a mouse model. 17 While MDM2 is a classic transcriptional target of p53, recent studies showed that MDMX is also a bona fide p53 target gene with its own (although weaker) p53-inducible P2 promoter.18,19 Therefore, both MDM2 and MDMX inducibility should be taken into consideration when analyzing and modeling the dynamics of the MDM2-negative feedback loop.
Ubiquitination of p53 by MDM2 and MDMX
MDM2 promotes p53 degradation by forming a stable complex through N-terminal domains. The MDM2 C-terminal RING domain recruits ubiquitin-conjugating enzyme E2 that performs covalent modification of p53 lysine residues.20,21 In addition to E3 ligase activity, MDM2 also interacts with the proteasome subunit C8 and may deliver substrates directly to the proteasome. 22 As expected, both the p53-binding domain and RING domains of MDM2 are critical for p53 degradation. However, the central acidic domain of MDM2 (residues 220-300) is also critical for ubiquitination of p53.23,24 The acidic domain has features of a partially unstructured region (Fig. 1A) that contains binding sites for most of the MDM2-binding proteins identified to date, including chromatin-modifying proteins (p300, YY1, KAP1, SUV39H1, EHMT1, etc.),25-27 de-ubiquitinating enzyme HAUSP, 28 ribosomal proteins, 29 and the tumor suppressor ARF. 30 Furthermore, the MDM2 acidic domain has been shown to bind weakly to the p53 core domain and cause a conformational change.31-35 This interaction may be important for exposing the target lysines in the p53 core for ubiquitination or orienting the 2 proteins for efficient ubiquitin transfer. 35 The disordered nature of the acidic domain probably provides the structural flexibility for interacting with multiple protein partners.36,37
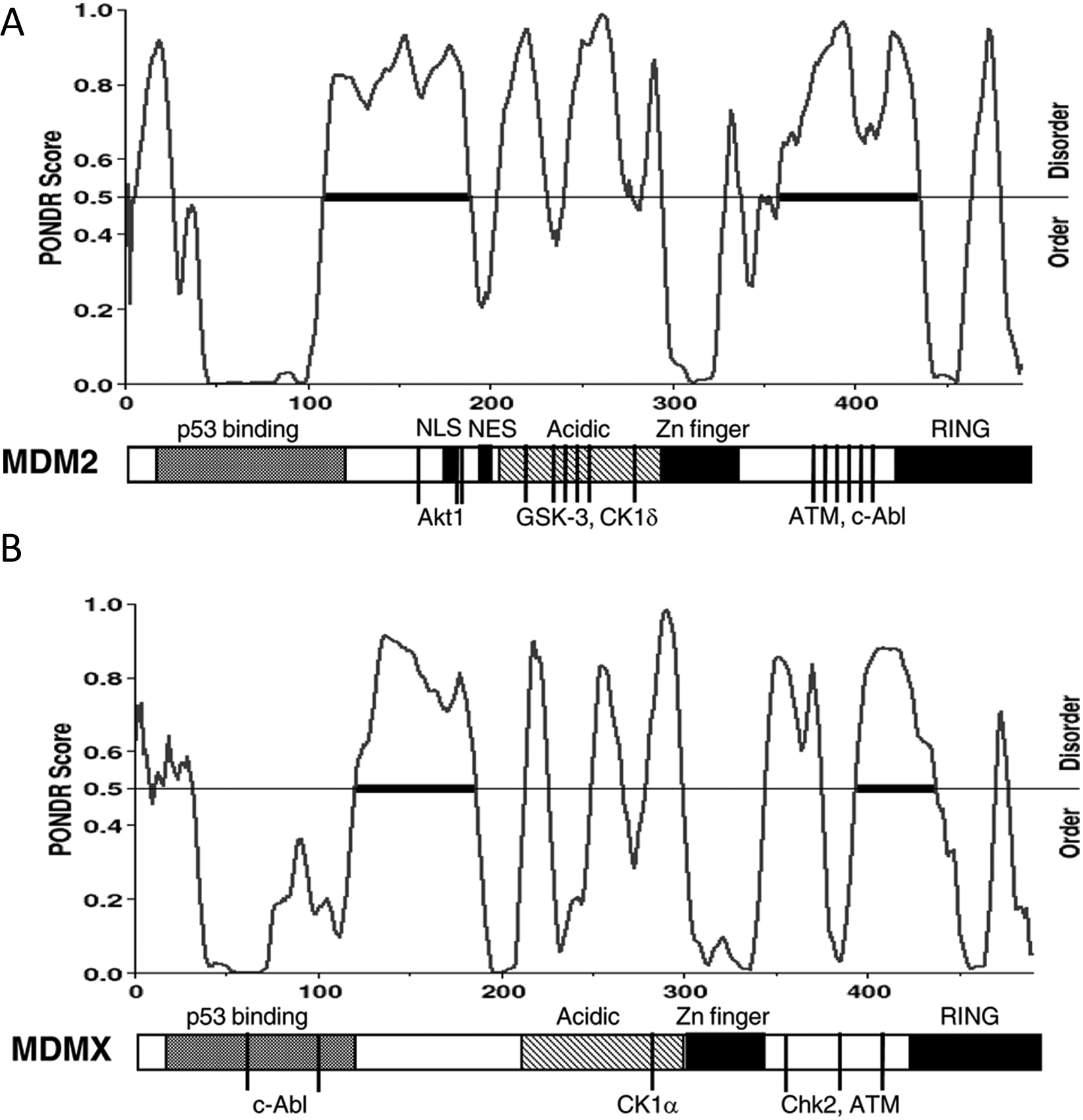
Diagrams of MDM2 (
MDMX has weak intrinsic E3 ligase activity. 38 The impact of MDMX expression on p53 stability is moderate in mouse embryonic fibroblasts (MEFs) derived from MDMX-null mice. 39 Similarly, knockdown or overexpression of MDMX in tumor cells generally causes little change in the p53 level. 40 Therefore, it has been proposed that MDMX regulates p53 mainly by formation of inactive p53-MDMX complexes.39-41 Early publications (pre-2004) showed that MDMX overexpression may even inhibit MDM2-mediated degradation of p53. It is noteworthy that some of these results may be due to the use of C-terminally epitope-tagged MDMX. Later work revealed that C-terminal epitope tagging is detrimental to MDMX ubiquitination by MDM2, suggesting an interference on MDM2 function. 42 The structural basis of this phenomenon was only apparent after more recent studies revealed the importance of the extreme C-terminal sequences of MDM2 and MDMX in RING domain dimerization and E3 ligase function.43,44
The fact that MDMX has a strong tendency to form heterodimers with MDM2 through RING domains, 45 the importance of RING domain dimerization for E3 activity, and the classic example of E3 ligase activation through BRCA1-BARD1 RING domain heterodimerization led many to hypothesize that MDM2-MDMX heterodimerization is important for p53 degradation. Indeed, biochemical experiments suggest that MDMX can stimulate the ability of MDM2 to ubiquitinate p53.46-50 The results of 2 recent mouse model experiments are consistent with a role of MDMX having at least a moderate role in regulating p53 stability in normal tissues. 51 Therefore, the relatively insignificant role of MDMX as a regulator of p53 stability in tumor cell lines may reflect abnormalities in the p53 pathway in transformed cells.
Stress Activation of p53
Many studies showed that different cellular stress and damage signals converge on MDM2 and MDMX to cause p53 activation. Oncogene-induced ARF expression induces p53 accumulation by binding MDM2 and inhibiting p53 ubiquitination. 52 Inhibitors of rRNA transcription (such as actinomycin D, 5-FU, and growth factor deprivation) induce ribosomal stress, which stimulates MDM2 interaction with several ribosomal proteins (such as L5, L11, and L23) that also block p53 ubiquitination. 53 These proteins interact with the MDM2 acidic domain, highlighting the importance of the acidic domain in sensing such growth-related stress signals.
DNA damage is probably the most extensively studied p53 activation signal. A critical player in p53 DNA damage response is the ATM kinase. 54 ATM is activated within minutes after DNA double-strand breaks and phosphorylates numerous substrates involved in cell cycle regulation and DNA repair.55,56 ATM activation of Chk2 kinase further amplifies the signal and expands the number of target proteins. Loss of ATM prevents the rapid accumulation of p53 after Ionizing radiation (IR) and abrogates p53-mediated cell cycle arrest response. 54 The importance of the ATM-mediated response is not limited to direct DNA damage by irradiation. A range of physiologically relevant stresses, including chemotherapy, oncogene activation, oxidative stress, and heat shock, can also trigger ATM activation either directly or by inducing DNA damage.
The Role of p53 Phosphorylation in DNA Damage Response
Although not the focus of this review, it is important to note that phosphorylation of p53 is an integral part of the signal transduction pathway that leads to its activation. MDM2-p53 binding has been extensively studied as a target of regulation by DNA damage. Several studies showed that DNA double-strand breaks induce phosphorylation of p53 S15 by ATM or DNA-PK.57,58 ATM also activates Chk2, which in turn phosphorylates p53 on S20, which is part of the MDM2-binding site.59,60 However, mutation of multiple phosphorylation sites including S15 and S20 does not abrogate p53 stabilization after DNA damage in cell culture.61-63 Mouse models showed that blocking p53 phosphorylation on S18 and S23 (equivalents of S15 and S20 in human p53) partially reduced p53 accumulation after DNA damage and caused partial defects in apoptosis and tumor suppression. 64 S23 single-site mutation also caused partial defects in p53 stabilization by gamma irradiation and increased the incidence of B-cell lymphoma. 65 Single-site mutation of S18 had no significant effect on p53 stabilization or tumor suppression but causes poor activation of certain p53 target genes after DNA damage. 66 These studies showed that p53 phosphorylation contributes to its stabilization but also implicate the presence of additional signaling mechanisms.
DNA Damage–Induced Phosphorylation of MDM2
Since p53 phosphorylation is not sufficient to mediate its stabilization after DNA damage, its E3 ligase MDM2 is a logical target for regulation. DNA damage has been found to induce MDM2 phosphorylation on serine 395 by ATM, 67 on serine 407 by ATR, 68 and on tyrosine 394 by c-Abl. 69 Phosphomimetic mutations of these sites inhibit MDM2-mediated degradation or nuclear export of p53.67,70 Recent mass spectrometric analysis of MDM2 purified from irradiated cells revealed the presence of additional phosphorylation sites (S386, T419, S425, and S429). Two of these sites were confirmed to be ATM targets and were strongly induced by DNA damage. 71 As expected for most phosphorylation sites, they cluster in a region of MDM2 that is disordered (Fig. 1A). 72 The MDM2 phosphorylation sites appear to have significant functional redundancy in regulating p53 degradation. Phosphomimetic substitution of a single site can strongly inhibit p53 degradation. 71 Substitution of all phosphorylation sites with alanine results in an MDM2 protein that continues to degrade p53 after DNA damage.
If DNA damage inhibits the E3 ligase activity of MDM2 through phosphorylation, the ubiquitination level of stabilized p53 is expected to be lower in damaged cells. The literature contains 2 conflicting conclusions in this regard using 2 different definitions of ubiquitination level. Some studies concluded that p53 ubiquitination level does not reduce or even increase after DNA damage. This is generally referring to the total level of ubiquitinated p53 by comparing identical amounts of cell extract, not taking into consideration that there are much higher levels of p53 in the damaged cells. However, the turnover rate of the entire p53 population is dictated by the ratio of Ub-p53/free p53, not the total level of Ub-p53 in the cell. In fact, when p53 loading was equalized to offset the stabilization effect, the fraction of p53 present in ubiquitinated form was significantly lower after DNA damage. 73 This is consistent with the observation that MDM2 purified from damaged cells has reduced E3 activity in ubiquitinating p53 in vitro. 73 These recent findings favor the conclusion that DNA damage inhibits p53 ubiquitination.
ATM Regulates MDM2 Dimerization
DNA damage signaling is highly efficient in regulating MDM2 function; even cell lines overexpressing MDM2 undergo p53 stabilization after irradiation. Therefore, ATM phosphorylation of MDM2 may be targeting structural features critical for its E3 ligase activity. Although the understanding of E3 ligases is still incomplete and rapidly evolving, certain common features have emerged as potential general targets for regulation. It appears that dimerization is often required for the activation of RING domain E3 ligases or the structurally similar U-box E3 ligase.74-78 E2-conjugating enzymes also frequently form dimers, both charged and uncharged with ubiquitin. 79 A secondary noncovalent ubiquitin-binding site on UbcH5 also allows the formation of oligomers, consisting of UbcH5 covalently charged with ubiquitin. 80 Dimerization and oligomerization by proteins in the ubiquitination pathway may be important for promoting the synthesis of polyubiquitin chains by increasing the local concentration of E2, providing a scaffold to access the end of a growing ubiquitin chain, and increasing the probability of ubiquitin-ubiquitin conjugation over ubiquitin-substrate conjugation.
The MDM2 C-terminal fragment containing the RING domain behaves as a high molecular weight oligomeric complex in gel filtration chromatography81,82 and can be cross-linked into dimers and oligomers by chemical cross-linking. 73 MDM2 phosphorylation by ATM or phosphomimetic substitution inhibits RING domain oligomer formation during gel filtration and blocks RING domain dimerization in cross-linking assays.71,73 These findings suggest that ATM-mediated phosphorylation inhibits MDM2 RING domain homodimerization and oligomerization, preventing the formation of a scaffold for synthesis of polyubiquitin chains on p53. Consistent with an important role of MDM2 oligomerization, artificially promoting MDM2 oligomerization in vivo by FKBP fusion or in vitro by GST fusion significantly stimulates the E3 ligase function of MDM2, particularly favoring the synthesis of long ubiquitin chains on p53.48,73
How does phosphorylation near the RING domain inhibit dimerization? Currently, the atomic structure of only the RING domain of MDM2 in isolation has been determined by nuclear magnetic resonance (NMR) and crystallography. The ATM sites located in a region adjacent to the RING domain that is unstructured thus were removed in structural studies in order to improve the solubility of recombinant proteins.43,83 It appears that the RING domain itself is sufficient for dimerization and even oligomerization.81,83 Recent analysis showed that inclusion of adjacent sequences reduces RING domain dimerization efficiency, suggesting that the ATM sites are located in a region that negatively regulates RING domain dimerization. 73 It is conceivable that after phosphorylation by ATM, the regulatory sequence adopts a conformation that can more efficiently conceal the dimerization surface of the RING domain through intramolecular binding, thus blocking dimerization.
Other Potential Effects of MDM2 Phosphorylation by ATM
In addition to direct regulation of RING domain dimerization, phosphorylation of MDM2 may also regulate interactions with other proteins. Several factors have been shown to cooperate with MDM2 in p53 polyubiquitination, such as p300/CBP and UBE4B.84,85 These proteins are thought to have E3 ligase activity of their own and interact with preformed ubiquitin conjugate on the substrate to further extend the ubiquitin chains. When recruited by MDM2, they may play a role in chain elongation on p53. The de-ubiquitinase HAUSP is an important regulator of p53 and MDM2 stability.86,87 MDM2 interaction with HAUSP is stimulated by DNA damage, which promotes p53 de-ubiquitination, 88 possibly also contributing to p53 stabilization.
DNA damage has been shown in many studies to cause transient down-regulation of MDM2 levels. Recent findings suggest that data related to this phenomenon should be treated with caution. This is due to the fact that many studies used SMP14 or 2A10 to detect MDM2 because of the availability and sensitivity of these antibodies. However, the 2A10 epitope on MDM2 contains S395 and can be masked by ATM-mediated phosphorylation.67,89 SMP14 reactivity to MDM2 is also blocked by phosphorylation of an unknown site in its epitope. In fact, it has been shown in a previous study, 90 and confirmed in recent experiments, that the MDM2 level does not undergo a significant decrease after irradiation when detected using other antibodies. 91 In certain publications, the MDM2 level decreased after high-dose ultraviolet (UV) irradiation (>20 J/m2) and was interpreted as protein degradation, disregarding the fact that high-dose UV strongly down-regulates MDM2 mRNA. 92 Therefore, whether MDM2 stability is regulated by phosphorylation remains to be demonstrated.
Phosphorylation of the MDM2 Acidic Domain Promotes p53 Degradation
While most of the phosphorylation sites stimulated by DNA damage are located near the C-terminal region of MDM2, the central acidic domain of MDM2 (220-300) also contains multiple serine phosphorylation sites that are constitutively modified in the absence of stress but down-regulated by DNA damage. 93 GSK3 and CK1δ kinases have been shown to phosphorylate these sites.94-96 Down-regulation of GSK3 levels by DNA damage may explain the reduction in acidic domain phosphorylation. 95 Alanine substitution of some of the phosphorylation sites in the acidic domain significantly inhibits p53 degradation without abrogating ubiquitination. A recent study suggests that the acidic domain phosphorylation sites regulate the ability of MDM2 to interaction with the 19S proteasome regulatory subunit, thus regulating MDM2 delivery of ubiquitinated p53 for degradation by the proteasome. 97
C-Terminal ATM Sites Regulate the MDM2 Acidic Domain
Deletion analysis showed that the MDM2 acidic domain is important for ubiquitination and degradation of p53.23,24 Acidic domain interaction with ARF and ribosomal proteins also inhibits p53 ubiquitination. 53 The mechanism by which the acidic domain participates in p53 ubiquitination remains poorly defined. Several groups showed that the MDM2 acidic domain binds to the p53 core domain.31,32,98 The binding affinity between the acidic domain peptides and the p53 core domain (Kd = 1,000 nM) is significantly weaker than the canonical N-terminal interaction (Kd = 130 nM) but may be important for stabilizing the complex and proper orientation of p53 for ubiquitination reactions to occur. 32 The acidic domain–p53 core interaction is also responsible for inducing conformational change in p53, which may help expose the target lysine residues in the core.34,35 As expected, CK1δ phosphorylation of the MDM2 acidic domain stimulates binding to the p53 core. 99 However, recent experiments showed that the acidic domain of MDM2 is also regulated by the C-terminal ATM sites. The weak binding between the acidic domain and the p53 core is inhibited by ATM after DNA damage, and the ability of MDM2 to induce p53 conformational change is also enhanced by ATM site mutations. 73 Therefore, the ATM sites control the conformation and functions of multiple domains on MDM2.
MDMX Phosphorylation by ATM, Chk2, and c-Abl
MDM2 undergoes self-ubiquitination and has a very short half-life in cell culture (15-30 minutes). In contrast, MDMX has a significantly longer half-life (>3 hours). The MDMX level is controlled by MDM2-mediated ubiquitination in a stress-dependent fashion.42,100,101 A fraction of MDMX from cells with DNA damage has delayed migration consistent with phosphorylation in an ATM-dependent fashion. 102 Candidate approach and mass spectrometry analysis have identified several phosphorylation sites near the C-terminal RING domain (S342, S367, S403).102,103 S342 and S367 were phosphorylated by Chk2, whereas S403 was modified by ATM (Fig. 1B). UV irradiation also induces S367 phosphorylation through activation of Chk1 kinase. 104 In vivo metabolic labeling experiments showed that S367 phosphorylation is the most prominent modification site. 105 This is consistent with the functional significance of S367 on MDMX ubiquitination: S367A substitution significantly reduced ubiquitination by MDM2, whereas S342A and S403A substitutions have negligible effects. 102 In addition to the C-terminal phosphorylation sites, it has been reported that the p53-binding domain of MDMX is phosphorylated by c-Abl on Tyr-99 and Tyr-55. Phosphorylation of Tyr-99 interferes with p53 binding, presumably facilitating the activation of p53. 106
MDMX Phosphorylation Controls Subcellular Localization
MDM2 has both Nuclear Localization Signal (NLS) (179-185, RQRKRHK) and Nuclear Export Signal (NES) (197-199, LSFDESLAL) sequences and undergoes nuclear-cytoplasmic shuttling. Akt1-mediated phosphorylation (S166, S186, S188) near the NLS has been shown to promote MDM2 nuclear translocation and enhance its ability to inactivate p53 in response to growth factor signaling.107-110 In contrast, MDMX appears to lack the NLS and NES signals in the corresponding locations, and phosphorylation in this region has not been reported. In the absence of stress, MDMX is predominantly cytoplasmic but shows strong nuclear accumulation after DNA damage. 111 Although nuclear proteins such as p53 and MDM2 can bind to MDMX and promote MDMX nuclear import, DNA damage–induced MDMX nuclear import also occurs in p53- and MDM2-null cells.111,112 It has been shown that phosphorylated S367 becomes a high-affinity docking site for 14-3-3, suggesting that 14-3-3 binding may promote conformational change in the RING domain to expose a cryptic NLS.104,105,113 Although MDMX nuclear translocation is a long-established phenomenon, the biological function of this shift remains speculative. It may serve to accelerate MDMX degradation because most of MDM2 is in the nucleus. Alternatively, it may be an active mechanism to suppress p53 activity in the nucleus.
MDMX Phosphorylation Promotes Ubiquitination by MDM2
A validated function of MDMX phosphorylation near the RING domain is to regulate degradation by MDM2. MDMX phosphorylation increases its binding affinity for MDM2, independent of nuclear import. In a cell free–binding reaction, GST-MDM2 binds to phosphorylated MDMX more efficiently than nonphosphorylated MDMX. 102 Transfection/Immunoprecipitation (IP) assay also showed that MDM2-MDMX co-precipitation efficiency is increased after DNA damage.114,115 Therefore, ATM-mediated phosphorylation of MDM2 near the RING domain inhibits homodimerization to suppress p53 ubiquitination, whereas Chk2-mediated phosphorylation near the MDMX RING domain promotes heterodimerization with MDM2 and enhances MDMX ubiquitination. Another effect of MDMX phosphorylation is the inhibition of MDMX-HAUSP binding, which may also contribute to increased MDMX ubiquitination.116,117 Dephosphorylation of MDMX by the WIP1 phosphatase also results in higher HAUSP binding and MDMX stabilization. 114
The biological relevance of MDMX phosphorylation sites has been verified in mouse knockin experiments. Alanine substitution of all 3 phosphorylation sites near the MDMX RING domain causes stabilization and resistance to DNA damage–mediated degradation in the MEFs and tissue of the mutant mice. 118 Furthermore, the animals have reduced p53 activation after irradiation and increased tumor incidence when introduced into a Myc transgenic background. These findings nicely mirror the cell culture results obtained mostly by overexpression and knockdown assays. Furthermore, p53 accumulation after DNA damage appears to be partially deficient in the 3A mice. 118 This suggests that MDMX phosphorylation may play a role in regulating p53 stability in vivo. This is consistent with the increased MDM2-MDMX heterodimer formation after DNA damage114,115 and p53 accumulation in MDMX RING domain mutant mice. 51
MDMX Phosphorylation by CK1α
MDM2 typically co-purifies with large amounts of ribosomal proteins (mainly L5, L11, and L23). In contrast, MDMX co-purifies mainly with 14-3-3 and near stoichiometric amounts of casein kinase 1 alpha (CK1α). CK1α interacts with the central region of MDMX including the acidic domain and zinc finger (150-350). S289 of MDMX has been identified as the major phosphorylation site by CK1α. 119 CK1α expression stimulates the binding between MDMX and p53 and leads to inhibition of p53 activity. As expected, pharmacological inhibition or knockdown of CK1α leads to p53 activation and cooperates with DNA-damaging drugs to activate p53 in culture. 119
Is endogenous CK1α important for regulating p53? CK1α is an abundant serine/threonine kinase that regulates multiple cellular pathways, such as Wnt/β-catenin signaling and circadian rhythm. 120 Circumstantial but supportive evidence has recently been reported in a mouse model with tissue-specific conditional knockout of CK1α in the intestine. 121 As expected from its role in the Wnt pathway, CK1α knockout leads to β-catenin stabilization and increased cell proliferation. CK1α knockout also leads to significant increases of p53 and p21 levels in vivo, which was interpreted as a secondary result of the proliferative stress from β-catenin stabilization. However, the data are also consistent with a role of CK1α through MDMX interaction. Mutation of the CK1α phosphorylation site on MDMX in mouse models will be needed to test the direct mechanism from CK1α.
Several questions need to be addressed in future studies. 1) How does CK1α binding and phosphorylation of the central domain of MDMX promote p53 binding? This effect implicates an interdomain communication between different parts of MDMX through conformational change or direct binding. 2) If the ATM sites on the MDM2 C-terminus can regulate the conformation and function of the acidic domain, it is possible that the ATM/Chk2 sites on the MDMX C-terminus also regulate acidic domain–CK1α binding. 3) The near stoichiometric binding between MDMX and CK1α is unusual for a kinase substrate relationship. This suggests that MDMX may also recruit CK1α to modify other proteins or regulate CK1α function.
Phosphorylation-Independent Mechanisms of MDMX Degradation
Interestingly, other types of stress that activate p53 without triggering DNA damage signaling also promote MDMX degradation. For example, ribosomal stress resulting from inhibition of rRNA transcription promotes MDMX degradation through L11-MDM2 interaction 40 ; oncogenic stress promotes MDMX degradation through ARF expression. 122 L11 binds to the zinc finger of MDM2 to promote MDMX ubiquitination, although detailed mechanisms remain undetermined. ARF also binds to MDM2 and stimulates ubiquitination of MDMX. A recent study showed that ARF promotes MDM2-MDMX heterodimer formation by mediating a second site interaction between the MDM2 and MDMX central domains. 122 This highlights the importance of MDM2 and MDMX homodimerization/heterodimerization as a common target for different stress signals, producing the same outcome of p53 stabilization and MDMX degradation.
Potential Intramolecular Interactions in MDM2 and MDMX
There is growing evidence that different domains of MDM2 and MDMX are functionally and allosterically connected. It has been suggested that p53 binding to the MDM2 N-terminus stimulates acidic domain–p53 core binding. This second site interaction is critical for p53 ubiquitination. 123 The ATM sites on the MDM2 C-terminus control both RING domain dimerization and acidic domain–p53 core interaction. 73 These findings suggest that the N-terminal p53-binding domain and C-terminal RING domain may communicate directly or indirectly. In fact, it has been shown that a point mutation in the MDM2 RING domain can cause conformational change in the acidic domain, which in turn increases p53 binding by the N-terminal domain. 124 There is also biochemical evidence that a C-terminal fragment of MDM2 binds weakly to the acidic domain, 125 suggesting the presence of an intramolecular interaction. MDMX also presents a similar picture: the ATM/Chk2 sites regulate RING domain heterodimerization and, by analogy to MDM2, may also regulate acidic domain function. CK1α binding to the acidic domain of MDMX promotes p53 binding, suggesting an allosteric connection to the N-terminal p53-binding domain.
Although static atomic structures of full-length MDM2 and MDMX may not be obtainable because some parts of these proteins lack rigid structures, it is likely that the flexible peptide sequences interact frequently with the stably folded domains and influence their conformation and function. In such a model, ligand binding or phosphorylation of one region may alter the intramolecular interaction between different domains. Such a mechanism plays a prominent role in the regulation of pRb-E2F1 binding by Cdk-mediated phosphorylation. 126 In the case of MDM2 and MDMX, intramolecular domain coupling may provide the structural basis for different signaling mechanisms to achieve an identical biological outcome, which is inhibiting p53 ubiquitination and promoting MDMX ubiquitination by MDM2.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health (CA109636 and CA141244).