
Abstract
The oncoprotein MDM2 is both the transcriptional target and the predominant antagonist of the tumor suppressor p53. MDM2 inhibits the functions of p53 via a negative feedback loop that can be circumvented by several ribosomal proteins in response to nucleolar or ribosomal stress. Stress conditions in the nucleolus can be triggered by a variety of extracellular and intracellular insults that impair ribosomal biogenesis and function, such as chemicals, nutrient deprivation, DNA damaging agents, or genetic alterations. The past decade has witnessed a tremendous progress in understanding this previously underinvestigated ribosomal stress-MDM2-p53 pathway. Here, we review the recent progress in understanding this unique signaling pathway, discuss its biological and pathological significance, and share with readers our insight into the research in this field.
Introduction
The p53 tumor suppressor is one of the most intensively studied proteins involved in tumorigenesis, principally because the p53 signaling pathway has been found to be defective in almost all human cancers. 1 As a homotetrameric transcription factor, p53 promotes the expression of a large number of genes that encode proteins directly responsible for p53-dependent cell cycle arrest, apoptosis, autophagy, senescence, and/or DNA repair. As a gatekeeper, p53, by directly or indirectly executing these cellular activities, is essential for maintaining genomic stability during cell growth and division and for preventing cancer. 2 Cancer cells can escape from p53 surveillance either by mutating its encoding gene, TP53, or by activating a number of proteins that suppress p53 activity. One of the predominant negative regulators of p53 (the other is MDMX 3 ) is the Ring-finger E3 ligase, MDM2. MDM2 and p53 participate in a negative feedback loop wherein p53 activates the transcription of MDM2,4-6 which in turn inactivates p53 by directly associating with it 7 and promoting its ubiquitination and proteasomal degradation.8-10
This MDM2’s antagonism of p53 can be circumvented by multiple cellular mechanisms in response to a multitude of stresses. DNA damage in a cell can lead to the inhibition of MDM2-mediated p53 degradation by the phosphorylation of MDM2 at multiple sites11-14 or by SCFβ-TRCP-mediated MDM2 turnover.
15
DNA damage signals can also cause the acetylation of p53 at specific lysine residues, which activates p53 by preventing MDM2-facilitated p53 turnover.
16
Oncogenic stress is another type of stress that can prevent MDM2’s inhibition of p53. It is often associated with the overexpression of the oncoproteins RAS or c-MYC. These 2 oncoproteins stimulate the expression of the tumor suppressor, the
The Emerging Ribosomal Proteins-MDM2-p53 Pathway
The ribosome is a complex multi-subunit machinery responsible for mRNA-to-protein translation. Ribosomal proteins, the individual subunits of this machinery, are synthesized in the cytoplasm and imported into the nucleolus where rRNAs, the other essential parts of this machinery, are synthesized (except for 5S rRNA, which is synthesized in the nucleoplasm) and processed. Subsequently, the 40s and 60s ribosomes are assembled separately in the nucleolus and exported into the cytoplasm to translate mRNA. 23 Disturbance of any single step in this process of ribosome biogenesis by distinct extracellular and/or intracellular insults can lead to nucleolar stress (also called ribosomal stress) and the consequent activation of p53 (Fig. 1).
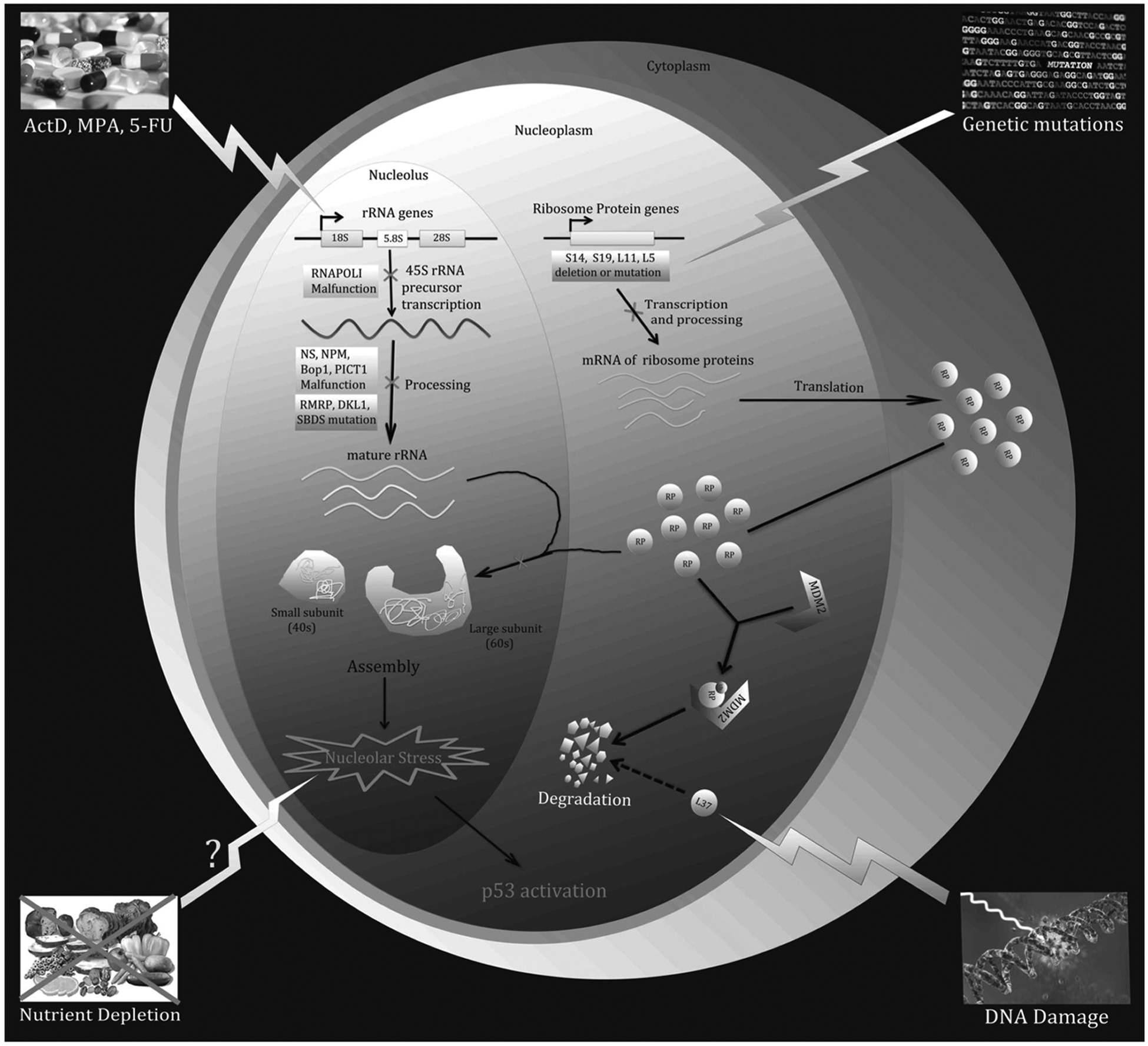
A variety of environmental reagents and genetic alterations can lead to nucleolar or ribosomal stress. Ribosome biogenesis involves 3 major events: (1) synthesis and processing of rRNA, (2) synthesis of RPs, and (3) assembly and cellular transportation of 40S and 60S ribosome units. Perturbation of each of these steps triggers nucleolar or ribosomal stress.
A variety of agents and signals have been shown to provoke nucleolar stress by impairing ribosome biogenesis. For instance, low doses of actinomycin D (Act D) (e.g., <10 nM), a commonly used anticancer drug, specifically inhibit RNA polymerase I and consequently prevent the transcription of rRNA,24,25 which leads to nucleolar stress. In addition to Act D, several chemical reagents have been found to trigger nucleolar stress by inhibiting rRNA processing or synthesis, such as 5-fluorouracil (5-FU)26-28 and mycophenolic acid (MPA). 29 Furthermore, it has been reported that overexpression of a dominant-negative mutant of Bop1, a nucleolar protein involved in rRNA processing, induces nucleolar stress. 30 Similarly, malfunction or deficiency in other nucleolar proteins required for rRNA production, such as Nucleostemin (NS), 31 PAK1IP1, 32 or hUTP18, 33 results in nucleolar stress. Interestingly, the tumor suppressor ARF, although known to activate p53 by preventing MDM2 from mediating the proteosomal degradation of p53, has been implicated in the inhibition of rRNA synthesis 34 by mediating the degradation of nucleophosmin (NPM1 or B23),35,36 blocking the phosphorylation of UBF1, 37 and preventing the nucleolar import of the RNA polymerase 1 transcription factor (TTF-1). 38 These studies suggest that ARF may also indirectly activate p53 by turning on the nucleolar stress pathway.
Increasing evidence suggests that nucleolar stress can be caused by an imbalance in RP levels 39 as the absence or malfunction of any RP will hinder the assembly of the 40S or 60S subunits. It has been shown that knockdown of 40S RPs, such as RPS6, 40 RPS14, and RPS19,41,42 leads to nucleolar stress and consequently p53 activation. The same phenomenon occurs when the 60S RPs, RPL29 and RPL30, are knockdown by RNAi. 43 Interestingly, DNA damage can induce nucleolar stress by inhibiting rRNA production 44 and increasing the proteasomal degradation rate of RLP37, which alters RP levels in the nucleolus. 45 Furthermore, genetic aberrations, including mutations and haploinsufficiencies in numerous RP genes, lead to impaired ribosomal synthesis and function. This is often associated with a range of clinical manifestations that are aptly named ribosomopathies. 46 For instance, Diamond-Blackfan anemia (DBA) is a ribosomopathy that is phenotypically characterized by a decrease in erythroid precursors. Patients diagnosed with DBA possess mutations in several different RP genes, including RPS19, RPS7, RPS10, RPS15, RPS17, RPS24, RPS26, RPS27A, RPL5, RPL11, RPL35A, and RPL36. 46 Haploinsufficiency of another RP-encoding gene, RPS14, causes a bone marrow dysfunctional phenotype called 5q-syndrome.47,48
Blocking the cellular transportation of RPs or ribosomal subunits prompts nucleolar stress. Nuclear import of RPs and export of preribosome are 2 key steps for 40S and 60S preribosome assembly and 80S ribosome assembly, respectively. It has been reported recently that siRNA-mediated depletion of IPO7 or XPO1 leads to nucleolar stress, eliciting p53 activation by blocking the cellular transportation of RPs and preribosome. 49 Intriguingly, this study also showed that these proteins are positively regulated by c-Myc but negatively regulated by p53, forming a regulatory feedback network, which is discussed in a later section.
In the early 1990s, it was reported that MDM2 and p53 can form a complex with 5S rRNA and RPL5. 50 Later on, nucleolar stress was shown to lead to p53 induction. 51 However, the link between the 2 seemingly irrelevant phenomena was not established until the physical and functional connections between RPs and the MDM2-p53 loop were demonstrated several years later.52-56 In these studies, RPL11, RPL23, and RPL5 were identified as proteins that prevent MDM2 from degrading p53 by directly binding to MDM2.52-56 The interaction of RPs with MDM2 in response to nucleolar stress is reminiscent of ARF’s interaction with MDM2 in response to oncogenic stress.17-21,57-60 The outcome of both the interactions is the same, as they ultimately bring about p53 activation and p53-dependent cell cycle arrest. Remarkably, the administration of Act D can trigger nucleolar stress pathways that stimulate the release of these RPs into the nucleoplasm, where they are able to associate with MDM2. This model is supported by the findings showing that siRNA-mediated ablation of either RPL11, RPL23, or PRL5 impedes Act D-elicited p53 activation in cultured cells.52-56
Since RPL11, RPL23, and PRL5 were initially discovered to play an ARF-like role in the inactivation of MDM2 and the activation of p53, more RPs, including RPS7,61,62 RPS3, 63 RPS27, 64 RPS27A, 43 and RPL26, 65 have been independently revealed by different laboratories to be involved in the MDM2-p53 regulation by binding to MDM2 and impairing its E3 ligase activity toward p53 upon nucleolar stress. Recently, our group demonstrated that RPS14, which is highly associated with 5q syndrome as briefly mentioned above and further discussed below, directly associates with the central acidic domain of MDM2 and prevents MDM2 from targeting p53 for degradation, which results in p53 activation and cell cycle arrest. 42 Moreover, we found that RPS14, like RPL11 and RPS7, is capable of regulating MDM2 stability independently of p53.
RPs can be divided into 2 subsets based on their ability to interact with MDM2. 66 The first subset includes MDM2-binding RPs that are able to activate p53 in response to nucleolar stress. This subset is referred as the “effector” RPs, including RPL11 and RPL5. The second subset consists of RPs that are unable to bind MDM2, such as RPS6 or RPS19. 66 Their depletion stimulates the “effector” RPs’ interaction with MDM2, consequently leading to the stabilization and activation of p53. Interestingly, RPS14 appears to belong to both of the 2 subsets. On one hand, nucleolar stress promotes the association of RPS14 with MDM2, which unties the MDM2-p53 loop. 42 On the other hand, ablation of RPS14 results in ribosomal stress and RPL11- and RPL5-dependent p53 activation. 42 This dual function feature has also been observed in several non-RP nucleolar proteins, such as nucleostemin (NS), PAK1IP1, and PICT1.31,32,67 An obvious question from these seemingly promiscuous studies is why cells need to use so many RPs to cope with MDM2’s negation of p53 activity when the nucleolar stress pathway is activated. In other words, are all of these recently revealed MDM2-binding RPs necessary for the nucleolar stress-MDM2-p53 pathway in vivo? This question is partially addressed by a genetic study that used a knockin mouse model system with a cancer-associated missense mutation MDM2C305F. 68 This MDM2 mutant was unable to interact with RPL11 and RPL5 in vitro 69 and in vivo. 68 Thus, the nucleolar stress activation of p53 was disabled in the MDM2C305F model system. This study firmly verifies the biological importance of the RP-MDM2 interaction in the nucleolar stress pathway. Since MDM2C305F could still bind to RPL23, this study also suggests that RPL23 might not be necessary for MDM2 inactivation in response to the nucleolar stress agents tested. 68 However, it is currently unknown whether MDM2C305F is able to associate with other RPs, such as RPS7 and RPS14, or how cells respond to the nucleolar stress independently of L11 and L5. Hence, there is little doubt now about the physiological existence of the nucleolar stress-MDM2-p53 pathway in cells and in animals (Fig. 2).
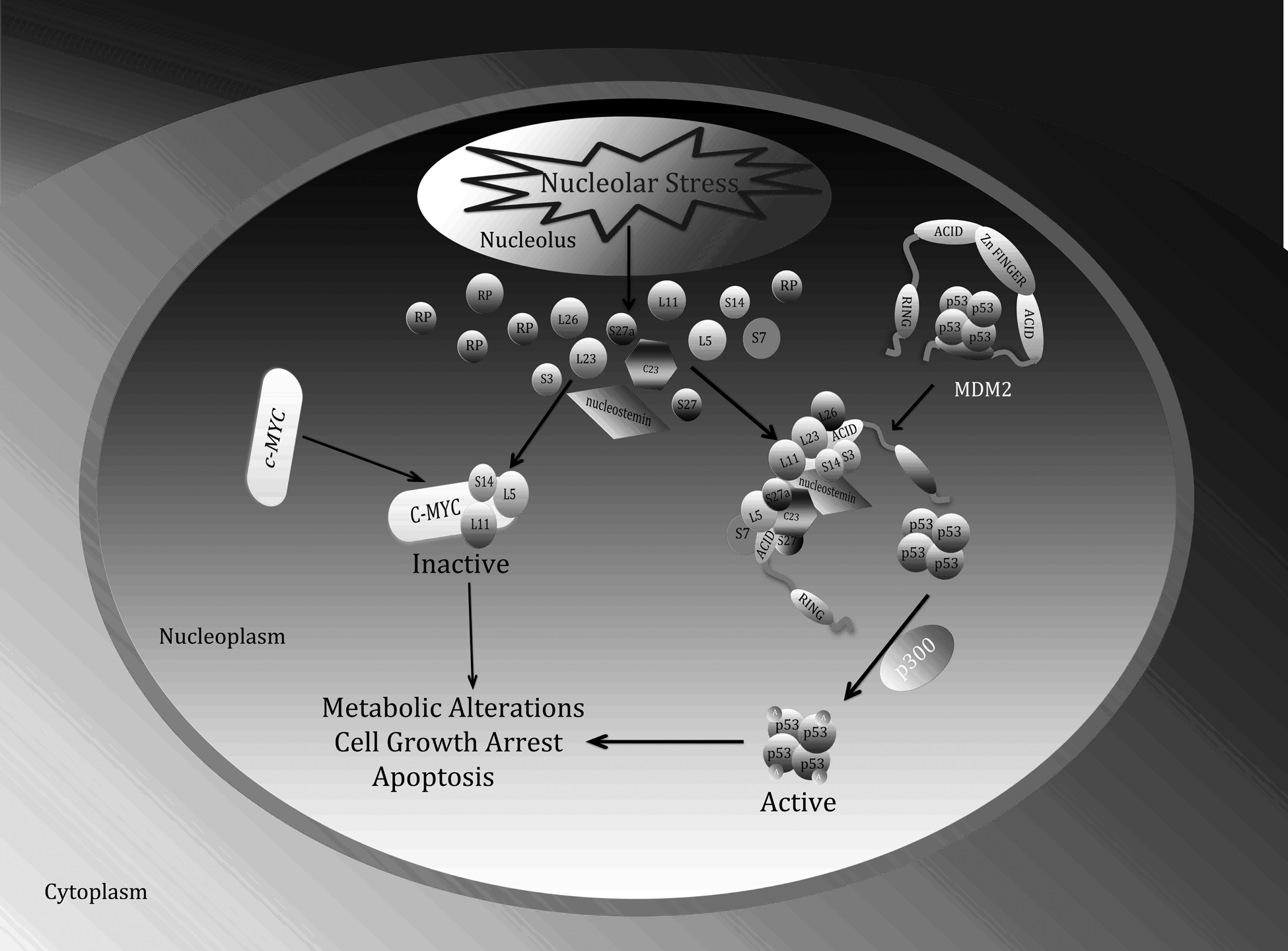
Interplay between the ribosomal stress-ribosomal proteins-p53-MDM2 pathway and the ribosomal stress-ribosomal proteins-c-Myc pathway. In response to nucleolar stress, several ribosomal proteins (RPs) as well as NS and C23 not only associate with MDM2 and inhibit its activity toward p53, consequently activating p53, but also suppress c-Myc transcriptional activity and c-Myc mRNA expression. As a concerted outcome, cells undergo growth arrest, senescence, and apoptosis.
New Insight into the Ribosomal Proteins-MDM2-p53 Pathway
Post-translational modifications of RPs
The discovery of the interaction of several RPs with MDM2 in response to nucleolar stress42,43,52-56,61-65 makes one wonder how this interaction is regulated. In other words, would modifications of MDM2-binding RPs play a role in further modulating this interaction and consequently the activation of p53? This appears to be the case at least with some of the MDM2-binding RPs. For example, a recent study showed that a subset of RPs, including RPL11, RPS7, and RPS3, undergo NEDD8-mediated modifications that protect them from degradation. 70 Another study showed that a prolonged lack of RP11 NEDDylation impairs RPL11’s ability to activate p53. However, the administration of Act D produces a rapid decrease in RPL11 NEDDylation, causing RPL11 to relocalize to the nucleoplasm where it activates p53 by binding to MDM2 and inhibiting its activity. 71 Interestingly, de-NEDDylated RPL11 molecules can be transiently recruited to the p53 responsive DNA promoter to enhance p53’s transcriptional activity. 72 Thus, NEDDylation offers a negative regulation to some MDM2-binding RPs in the ribosomal stress-MDM2-p53 pathway.
By contrast, the regulation of the MDM2-p53 pathway through the ubiquitination of certain MDM2-binding RPs seems more sophisticated. Several RPs, such as RPL26, RPS27A, and RPS7, have been reported to be ubiquitinated by MDM2.43,61,73 Of these RPs, RPL26 and RPS27A are ubiquitinated by MDM2 for proteosomal degradation under nonstressed conditions. When stress occurs in the cell, MDM2’s effect on RPL26 and RPS27A is mitigated and the 2 proteins are free to enhance the cellular expression of p53. Thus, RPL26 and RPS27A are important parts of the MDM2-p53 regulatory loop.43,73 In contrast to the regulation of the 2 RPs by MDM2, RPS7 is not targeted for proteosomal degradation when this RP is ubiquitinated by MDM2. Instead, ubiquitinated RPS7 prolongs p53 stabilization and activation and leads to p53-dependent apoptosis, which is one extended consequence of nucleolar stress. 61
Usually, nucleolar stress preferentially elicits p53-dependent cell cycle arrest, but not apoptosis, as this is a type of nongenotoxic stress and affected cells would not need to be eliminated through apoptosis. 42 However, some nucleolar stress also induces apoptosis via RP-dependent activation of p53. This difference can be partly explained by the following studies. Recently, RPL11, RPS7, RPS27A, and RPS14 have been shown to stabilize MDM2 independently of p53.42,43,61,74 As a result, these accumulated RP-associating MDM2 molecules directly bind to p53 and suppress its transcriptional activity instead of mediating p53 ubiquitination and degradation, as the MDM2-E3 ligase activity toward p53 is impaired by these RPs upon nucleolar stress. Under this type of control by MDM2, p53 may only selectively bind to a subset of p53 responsive DNA promoters, including p21 and MDM2, but not to the promoters in the genes that are essential for apoptosis. This difference in promoter selection by p53 may also be due to the differences in its target promoter sequences and local environments. This difference happens via other modifications of p53 as well. For example, the acetylation of different lysine residues within p53 guides acetylated p53 molecules to different promoters 75 ; in addition, loss of p63 and/or p73 impairs p53-dependent transcription of only a subset of p53 target genes. 76 Similar to those occasions, the increase of only RPS7’s steady state levels can induce p53-dependent cell cycle arrest, whereas the elevation of ubiquitin-modified RPS7 molecules is capable of eliciting apoptosis, as the former form of RPS7 could only activate the expression of p21, and the latter could induce the expression of those proapoptotic p53 target genes, such as Bax or Puma. In this regard, MDM2-mediated ubiquitination, but not degradation, of RPs may assist p53 in selecting its target promoters in response to nucleolar stress. This broader promoter selection allows RPS7-activated p53 to robustly activate the expression of not only p21 but also other proapoptotic target genes. Detailed mechanisms underlying this complex regulation of p53’s promoter selection remain largely obscure.
In addition to NEDD8- and Ubiquitin-conjugating modifications, RPL5, RPS3, and RPS14 are post-translationally modified by phosphorylation.77-80 However, it is unknown whether these phosphorylation events are connected to the nucleolar stress pathway and the regulation of MDM2. RPS6 is also post-translationally modified by phosphorylation. Experimental evidence suggests that the phosphorylation of RPS6 may be associated with the translational control of TOP mRNAs, glucose metabolism, and cell proliferation. 81 Although RPS6 deficiency has been shown to activate p53 and induce p53-dependent cell cycle arrest by impairing the 40S ribosome assembly through an RPL11-dependent mechanism, 40 the direct role of RPS6 phosphorylation in the MDM2-p53 feedback loop is still an open question.
Thus far, little attention has been paid to the possible participation of acetylation in regulating RPs’ functions in the MDM2-p53 pathway. A gross proteomic analysis of nucleolar proteins detected acetylation of many RPs, including RPL5, RPL11, RPL26, RPS3, RPS7, and RPS27A. 82 This preliminary finding suggests that acetylation of RPs may play a regulatory role in ribosome biogenesis and/or other extraribosomal functions, such as modulating the MDM2-p53 pathway. This is certainly an interesting and important question for future exploration.
Several new players in the RP-MDM2-p53 pathway
When talking about MDM2, it is impossible not to mention its close friend and partner, MDMX. MDMX was originally identified as an MDM2 homolog. 3 Subsequent studies have demonstrated that MDMX plays an almost equally important role as MDM2 in controlling p53 stability and activity.1,83,84 MDMX and MDM2 interact with each other through their C-terminal RING domains to promote the ubiquitination and proteosomal degradation of p53.85-87 Initial evidence showed that RPL11, RPL5, and RPL23 were unable to bind MDMX. However, several later studies have shown that the RP-MDM2-p53 pathway engages MDMX.22,88,89 First, nucleolar stress induces RPL11-MDM2 interaction, which facilitates MDM2-dependent MDMX degradation, thus leading to p53 activation. 88 Also, overexpression of MDMX confers Act D or 5-FU resistance and impairs nucleolar stress-induced p53 activation, although the stabilization of p53 is not affected. Furthermore, MDMX is required for the RP-mediated inhibition of MDM2 auto-ubiquitination. 61 In addition, co-expression of MDMX and RPS7 results in remarkably greater stabilization of MDM2 than does the expression of either MDMX or RPS7 alone. 61 Finally, 5S rRNA was recently found to associate with MDMX and protect it from degradation by MDM2. 90 This association was impaired upon nucleolar stress, leading to MDMX degradation by MDM2. 90 Altogether, these studies strongly demonstrate that deregulation of MDMX is also crucial for p53 activation upon nucleolar stress.
Other nucleolar proteins besides RPs participate in the RPs-MDM2-p53 pathway. A recent study uncovered PICT1 as one of the key regulators of this pathway. 67 PICT1 is encoded by a gene that resides on human chromosome 19q13.32. This region on chromosome 19 is called the tumor-suppressive region because it is frequently mutated in cancer cells. PICT1 was originally regarded as a tumor suppressor, as microarray data showed that there is a correlation between high malignancy rates and low expression levels of PICT1 in diffuse gliomas and ovarian cancers.91-93 Also, the ectopic expression of PICT1 in human glioma cells led to the stabilization of the tumor suppressor PTEN and promoted apoptosis.94-96 However, other studies suggested that PICT1 might not fit the canonical definition of a tumor suppressor. Patients with pure oligodendrogliomas with PICT1 haploinsufficiency have a statistically significant prolonged survival time.97-99 This conflict was resolved in a recent study that generated Pict1–/– mice. 67 The Pict1–/– mice in this study died during embryogenesis at day 3.5 (E3.5) because of aberrant apoptosis. When using Pict1–/– murine ES cells with exogenous Doxycycline-inducible Pict1, it was found that Pict1 deficient cells arrested at G1/S and underwent apoptosis due to the accumulation of p53. At a molecular level, PICT1 deficiency in ES cells promotes the interaction of several RPs with MDM2 including RPL11, RPL5, RPL23, and RPS7. The mechanism is reminiscent of the nucleolar protein NS, knockdown of which also prompts the interaction of RPL11 and RPL5 with MDM2 and inhibition of MDM2 by RPL11 and RPL5. 31 However, unlike the case of NS, 31 only the knockdown of RPL11, but not PRL5, PRL23, or PRS7, attenuated the activation of p53 in PICT1 deficient cells. This observation revealed that the interaction of Pict1 with RPL11 is essential for inhibiting MDM2. Confocal microscope experiments showed that PICT1 retains RPL11 in the nucleolus, where the latter is unable to interact with MDM2. In PICT1 deficient cells, RPL11 is free to relocate to the nucleoplasm where it binds to MDM2 and inhibits its E3 ligase activity toward p53. Clinical data also support the idea that PICT1 supports tumor progression. Patients with TP53-intact colorectal tumors and a low level of PICT1 expression had a higher 5-year survival rate than patients with high levels of PICT1 expression. 67 This enchanting study not only suggests that RPL11 can act as a tumor suppressor in wild-type p53-containing human cancers but also makes the PICT1-RPL11 interaction a potential target for anticancer drug discovery. A more recent report showed that in response to nucleolar stress, PICT1 directly interacts with p53 and stabilizes it. 100 This conflicting result makes the role of PICT1 in the regulation of the RP-MDM2- p53 pathway more sophisticated and intriguing.
One early discovered and well-studied nucleolar tumor suppressor protein is ARF.57,58 ARF’s classification as a tumor suppressor protein is primarily due to the ability of this protein to activate p53 in response to oncogenic stress.57,58 As discussed above, certain RPs also activate p53 by inhibiting MDM2. However, the activation of the MDM2-inhibitory functions of these RPs is solely in response to nucleolar stress, but not oncogenic stress. Since ARF impairs rRNA processing 34 by binding to B23 and mediating its degradation, blocking UBF1 phosphorylation, and preventing the nucleolar import of TTF-1, 38 it remains likely that high levels of ARF may also trigger the nucleolar stress pathway by engaging in crosstalk with some of the RPs involved in the MDM2-p53 feedback loop. Indeed, one of our recent studies verified this possibility as we demonstrated that ARF and RPL11 directly bind to each other in vitro and ex vivo. 101 This interaction was enhanced in response to either oncogenic or nucleolar stress. 101 Consistently, these 2 nucleolar proteins work as partners to form a complex with MDM2 and inhibit its activity toward p53. RNAi-mediated knockdown of RPL11 impaired ARF activation of p53. The simultaneous overexpression of ARF and RPL11 enhanced their ability to activate p53. 101 High levels of ARF increased the non-ribosome-associated form of RPL11 and enhanced the RPL11-MDM2 interaction, thereby heightening the ability of RPL11 to suppress MDM2 activity and consequently activate p53. 101 Although some questions remain to be addressed regarding the regulation of the RP-MDM2-p53 pathway by other non-RP nucleolar proteins, the studies as discussed above consolidate the concept that ribosomal proteins, such as RPL11, similar to ARF, also play a role in preventing neoplasia and tumor development by activating p53.
Chemical nature of RPL11-MDM2 interaction
The role of MDM2-binding RPs has been established in the regulation of the MDM2-p53 loop. However, the mechanism by which these RPs negate the inhibitory activity of MDM2 toward p53 remains largely unclear. To address this issue, several laboratories have tried to determine the molecular anatomy of the RP-MDM2 interaction. Interestingly, in a manner similar to ARF,57,58 all of the known MDM2-binding RPs appear to inactivate MDM2-E3 ligase activity by binding to the central region (encompassing the acidic and zinc finger domains) of this p53 ubiquitin ligase.42,43,52-56,61-65 This finding supports a previous study, confirming the importance of the central region in mediating p53 degradation. 102 Surprisingly, only RPL11 appears to bind to the zinc finger domain of MDM2, whereas other MDM2-binding RPs seem to bind to the central acidic domains adjacent to the zinc finger domain.42,43,52-56,61-65 The cancer-associated mutation C305F in the zinc finger domain of MDM2 failed to bind to both RPL11 and RPL5,68,69 but the C305S mutant MDM2 only failed to bind to RPL11. 88 This difference in binding raises the question of whether RPL11 or RPL5 binds to the zinc finger of MDM2 directly or whether mutating any of the zinc-chelating cysteine residues in this domain would cause a conformational change of the protein’s local structure, thus disabling MDM2 interaction with RPL5, RPL11, or possibly other RPs. This question was partially addressed in our recent study. 103 By using a set of biochemical, proteomic, mutagenic, biophysical, and cell-based assays, we found that the C4 zinc finger domain of MDM2 is essential for its direct contact with RPL11, but not RPL5. We also identified several noncysteine acidic or polar residues in the C4 zinc finger domain of MDM2 as well as several basic residues within RPL11, which were critical for their interaction with each other. 103 These results reveal that the interaction between RPL11 and MDM2 likely occurs through electrostatic interactions, such as H-bonding. However, whether these residues would make direct contact with each other as predicted in our study 103 will need to be confirmed by solving the crystal structure of the MDM2-RP complex in the near future. Interestingly, we also found that a single point mutation of the 4 cysteine residues within the zinc finger domain of MDM2 causes a tertiary, but not secondary, conformational change in its central domain and consequently impairs its association with RPL11 as well as RPL5 and RPL23. 103 Therefore, RPL11-binding to the zinc finger domain of MDM2 is important in negating MDM2 activity. Since MDM2 mutations often occur in its zinc finger domain in some human cancers, mutating any of the 4 cysteine residues of this domain would also interfere with the interaction of MDM2 with other RPs in addition to RPL11. 103 Although C305S can still bind to RPL5 but not RPL11, 88 this mutant might not be able to bind to other MDM2-binding RPs, such as RPL23, RPS7, RPL26, RPS27, RPS3, or RPS14. Altogether, these findings emphasize the essential role of the zinc finger domain of MDM2 in sensing nucleolar stress and also reveal the noncysteine hydrophilic residues within this domain as crucial amino acids for the RPL11-MDM2 binding. Therefore, the zinc finger domain of MDM2 could be a useful target for future anticancer drug discovery.
Regulation of ribosomal biogenesis by MDM2 and p53
A growing body of evidence suggests that MDM2 and p53 might also regulate ribosome biogenesis, potentially causing nucleolar stress in addition to being regulated in response to this type of stress (Fig. 3). Although initial findings showed that RPL11, RPL5, and RPL23 are not ubiquitinated or degraded by MDM2,52-56 later studies indicated that other RPs, such as RPS7 61 and RPS14 (our unpublished observation), are ubiquitinated by MDM2. The MDM2-mediated ubiquitination of these 2 RPs might be involved in regulating their function, as ubiquitination of RPS7 or of RPS14 by MDM2 does not result in proteosomal degradation. 61 By contrast, MDM2 mediated the degradation of RPL26, 73 RPS27, 64 and RPS27A. 43 In addition to these RPs, MDM2 can also mediate the degradation of RNA Pol I transcription termination factor, TTF-I, 104 which is essential for rRNA synthesis and processing. Therefore, high expression of MDM2 may trigger nucleolar stress, either by decreasing RP levels or by impairing rRNA biogenesis. The ability of MDM2 to induce nucleolar stress may account for its ability to suppress cell proliferation in a p53 independent manner. 105 This unorthodox concept has been supported by 2 additional studies showing that the overexpression of MDM2 in some cancer cells induces cell cycle arrest.106,107
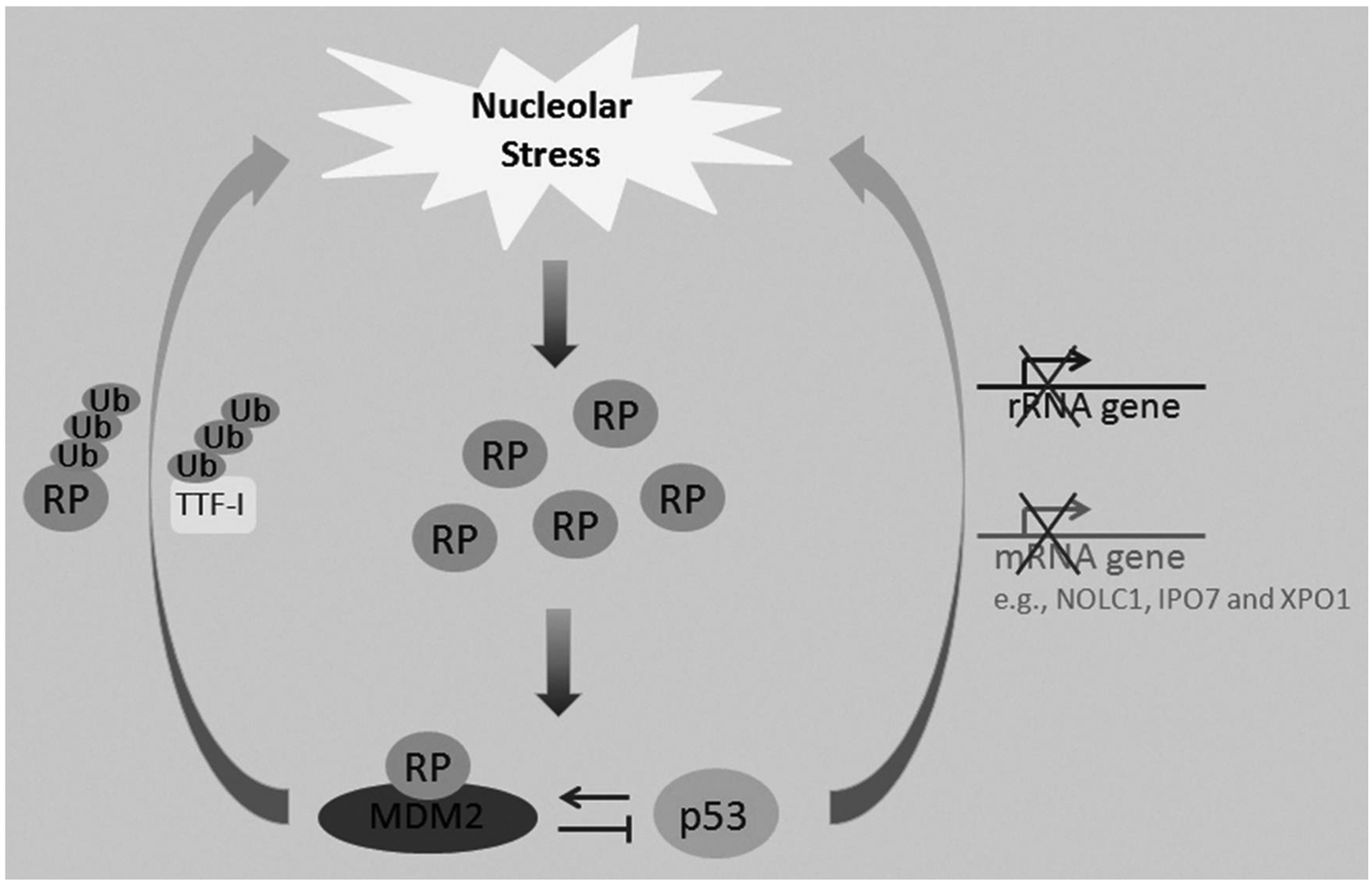
The nucleolar stress-MDM2-p53 regulatory circuit. Although nucleolar stress leads to p53 activation, recent studies also show that elevated p53 or MDM2 levels may trigger nucleolar stress.
Similar to MDM2, p53 can also induce nucleolar stress. It has been shown that p53 can impair ribosomal biogenesis by repressing RNA polymerase I transcription108,109 and thus reducing rRNA synthesis. Conversely, rRNA synthesis is significantly elevated in most human tumors with mutated p53. 110 By inhibiting the expression of an assembly chaperone, NOLC1, p53 has been found to impair small nucleolar ribonucleoprotein (snoRNP) assembly. 111 In addition, p53 transcriptionally represses the expression of IPO7 and XPO1, which are responsible for the cellular transportation of RPs and the pre-ribosome. 49 Collectively, these studies suggest that p53 may inhibit ribosomal biogenesis through multiple mechanisms to elicit nucleolar or ribosomal stress, which further stimulates p53 activity in a positive feedback fashion (Fig. 3).
As shown in a genetic study using Eµ-Myc/+ mice and RPL24- or RPL38-haploinsufficient mice, 112 elevated ribosome function and protein synthesis favored cancer cell proliferation, denoting the biological significance of the down-regulation of ribosomal biogenesis by p53. A number of proteins involved in ribosomal biogenesis and protein translation are transcriptionally regulated by c-Myc.113-115 The Myc oncoprotein has also been shown to augment protein synthesis, accelerating cell cycle progression and leading to cancer initiation independently of its transcriptional activity. 112 When crossing the Eµ-Myc/+ and RPL24- or RPL38-haploinsufficient mouse lines, tumors grew much slower in the hybrid mouse line than in Eµ-Myc/+ mice that harbored normal copies of RPL24 or RPL38. The oncogenic activity of Myc was markedly reduced due to haploinsufficiency of either RPL24 or RPL38; therefore, sufficient ribosomal biogenesis and protein production are necessary for tumor growth. This finding also suggests that the loss of one copy of either the RPL24 or RPL38 gene might cause nucleolar stress, leading to p53 activation and thus inhibiting Myc-driven tumorigenesis in the hybrid animals. Alternatively, haploinsufficiency of RPL24 or RPL38 might also reduce ribosome assembly, thus releasing more ribosome-free RPs into the nucleoplasm where they can inhibit c-Myc-dependent transcription (Fig. 2). Previous and recent studies have shown that RPL11, 116 RPL5, and RPS14 (our unpublished results) can repress c-Myc’s transcriptional activity by directly binding to it 116 or through a microRNA-mediated mechanism. 117 Hence, negative regulation of ribosome biogenesis by p53 and perhaps by MDM2 as an effector of p53 in this case inhibits tumor growth, whereas positive regulation of ribosomal biogenesis by c-Myc enhances tumor growth.
The Double-Edged Sword
As discussed above, one obvious biological outcome of turning on the nucleolar stress-RPs-MDM2-p53 pathway is the cessation of cell proliferation and growth, thus likely slowing down or preventing tumor growth. 22 In support of this statement, a number of cell-based assays have consistently demonstrated that in response to Act D or 5-FU induced nucleolar stress, each of the known MDM2-binding RPs relocalizes to the nucleoplasm and prevents MDM2-mediated degradation of p53, inducing p53-dependent cell cycle arrest and growth inhibition.42,43,52-56,61,62,64,65 Further supporting the hypothesis that RP-mediated p53 may have an antitumorigenic effect is an elegant study published recently using MDM2C305F knockin mice. 68 Since MDM2C305F was unable to bind to RPL11 and RPL5,68,69 the 2 RPs failed to inactivate MDM2 and to activate p53 in MDM2C305F knockin mice when the animals were treated with either Act D, 5-FU, or MPA. 68 Due to this failure, MDM2C305F/Eµ-Myc/+ mice developed c-Myc-driven lymphomagenesis more rapidly than did the animals with the transgenic Eµ-Myc/+ only. 68 The anticancer function of RPL11 was further validated by the other animal study described above on the role of PICT1 in cancer development, 67 as PICT1 was found to retain RPL11 in the nucleolus and the reduced level of PICT1 resulted in the release of RPL11 to the nucleoplasm, where the latter bound to MDM2 and inhibited its activity, leading to p53 activation. 67 Also lower PICT1 levels were associated with lower incidence of human colorectal or esophageal cancers that harbor wild-type p53. 67 Clearly, the anticancer function of the RP-MDM2-p53 pathway is beneficial to human health.
However, the inappropriate activation of this pathway can have pathogenic effects on cell and tissues. Human bone marrow tissues seem to be particularly sensitive to the aberrant expression of p53 via the nucleolar stress pathway. 46 The tissue specificity of these ribosomopathies may be the result of the intense need for ribosome biogenesis in proerythroblasts. Diamond-Blackfan anemia (DBA) is a congenital disease that is caused by a reduced rate of erythrocyte differentiation in the bone marrow. Phenotypically, DBA is characterized by red blood cell aplasia, macrocytic anemia, clinical heterogeneity, and increased risk of malignancy.46,118 Interestingly, this genetic disease is highly associated with mutations of several RP-encoding genes including RPS19 (25%), RPL5 (19%-21%), and RPL11 (7%-16%).119-121 This association is supported by mouse models that conditionally express Rps19. In these experiments, Rps19 deficient mice developed a phenotype similar to that of human DBA, 122 as they suffered from macrocytic anemia and leukocytopenia. 122 At the molecular level, the impairment in ribosome biogenesis leads to activation of the nucleolar stress pathway, which aberrantly activates p53 and causes a reduction in bone marrow cell proliferation.46,122 Another human disease that is caused by impaired ribosome biogenesis is 5q-syndrome, which often occurs in adult females and is characterized by macrocytic anemia, often erythroblastopenia, thrombocytosis, and megakaryocyte hyperplasia with nuclear hypolobation. 46 Patients with this genetic disease tend to have interstitial deletions within a region on chromosome 5q that contains Rps14.47,48 The mapping of 5q syndrome to Rps14 has been validated by the generation of an animal model that conditionally knocks out the Rps14 gene, as Rps14 deficient mice phenotypically resemble 5q-syndrome. 47 Remarkably, deletion of the TP53 gene in the 5q-syndrome mice completely rescued the progenitor cell defect, restoring common myeloid, megakaryocytic-erythroid, and granulocyte-monocyte progenitors as well as hematopoietic stem cell bone marrow populations. 47 Thus, DBA and 5q syndrome are both caused by the inappropriate activation of p53 during bone marrow development. 122 However, 5q syndrome is the result of RPS14 haplodeficiency, 47 whereas DBA is the result of mutations in RPS19, RPL5, or RPL11. Since these genetic myelodysplastic syndromes (MDS) are all caused by the abnormality of ribosome biogenesis, DBA and 5q syndrome are therefore classified as ribosomal diseases or ribosomopathies. 46 Not only the mutations of the above mentioned RPs but alterations of other genes, such as SBDS, DKC1, RMRP, TCOF1, and NPM/B23, involved in rRNA synthesis and/or processing have been reported to cause ribosomopathies, leading to increased incidences of cancers. 46 These human genetic studies have been recapitulated in both murine and zebrafish model systems. Mice with haploinsufficiency of Tcof1, the murine homolog of the Treacher Collins syndrome (TCS)–responsible gene, developed a TCS-like phenotype, which could also be eliminated by inhibiting p53 activity in the animals. 123 Several zebrafish models of DBA and other ribosomopathies have been generated as well. Defective hematopoiesis and other developmental abnormalities (e.g., brain or craniofacial defects) were found in the zebrafish mutants or morphants (morpholino-mediated gene down-regulation) of RP genes, such as rpl11, rps19, rps7, or rps29.124-129 Again, most of the phenotypes due to these RP mutations are at least partially, if not completely, rescued by further down-regulating p53 levels in the zebrafish models. Interestingly, in addition to p53, induction of deltaNp63 in the nonneural ectoderm and blood cell progenitors was found to contribute to the craniofacial and erythroid defects in the rps19-deficient morphants, 126 suggesting that the entire p53 family members including p63 and possibly p73 might be involved in the nucleolar stress response and its associated ribosomopathies. Although these studies demonstrate that p53 induction upon nucleolar stress promotes extensive apoptosis in certain progenitor cell types leading to DBA or other ribosomopathies, the reason why the patients with these diseases have a high incidence of malignancies remains elusive.
In summary, the activation of the RP-MDM2-p53 pathway is a double-edged sword to human health (Fig. 4). On one hand, the RP-mediated inactivation of MDM2 and activation of p53 in the nucleolar stress pathway prevent neoplasia and tumor development. This anticancer effect is clearly conducive to human health. On the other hand, the inappropriate activation of the RP-MDM2-p53 pathway during bone marrow development can be pathogenic to normal progenitor cells and tissues. The aforementioned ribosomopathies, DBA and 5q syndrome, are unfortunate outcomes of this malfunction. Therefore, further dissection of this pathway at the atomic and molecular level will offer more insight into the detailed mechanisms underlying these human genetic diseases and provide information that will help identify potential molecular targets for future anticancer drug discovery.
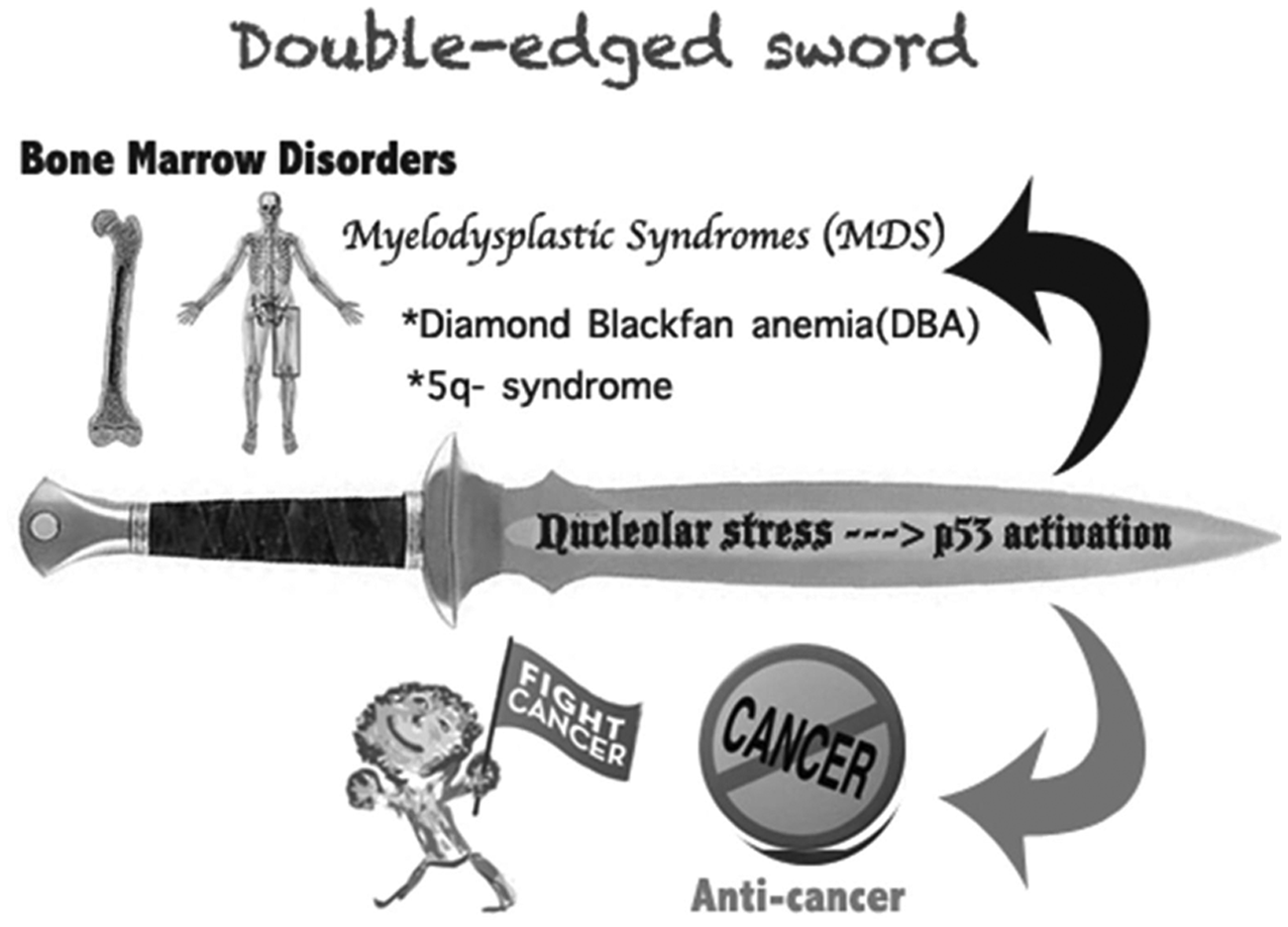
A double-edged sword: the nucleolar stress-p53 activation. The RP-mediated p53 activation by inactivating MDM2 upon nucleolar stress causes a dual effect on human health. On one hand, p53 activation prevents or retards tumor growth in response to nucleolar stress; on the other hand, inappropriate p53 activation due to ribosome dysfunction leads to myelodysplastic syndromes (MDS), such as DBA or 5q syndrome.
Questions and Prospects
Much has been learned about the recently acknowledged nucleolar stress-p53 signaling pathway and its relevance to human cancers and genetic diseases. However, there are still a lot of missing pieces in this puzzle. One large remaining issue that was mentioned above is how the MDM2-binding RPs deactivate MDM2-E3 ubiquitin ligase. There are currently 2 known facts regarding this regulation: (1) multiple RPs have been shown to inhibit MDM2 activity by directly binding to this protein,42,43,52-56,61-65 and (2) RPL11 directly binds to the zinc finger domain of MDM2 while the other MDM2-binding RPs interact with its central acidic domain.52,53,103 Hence, this large question can be divided into the following specific questions. First, where do these RPs interact with MDM2: in the nucleoli or in the nucleoplasm? It seems that this question has already been addressed, as several studies have repeatedly shown that RPs, such as RPL11 or RPL5, are released from the nucleolus to the nucleoplasm, where they bind MDM2.52,53,55 Also, the overexpression of RPs such as RPS14 does not affect the interaction of MDM2 with p53. 42 RPL11 was recently shown to potentiate MDM2-mediated degradation of nucleoplasmic MDMX, 88 suggesting that RPs do not appear to relocalize MDM2 to the nucleoli. The second specific question is whether MDM2-binding RPs cooperate with one another to synergistically inactivate MDM2. This appears to be the case, as RPL11 and RPL5 have been shown to work together via 5S rRNA to inhibit MDM2. 130 Furthermore, in transgenic animal studies, these 2 RPs are unable to bind to MDM2C305F and activate p53 in response to nucleolar stress. 68 However, it is still unclear whether RPL11 cooperates with other known MDM2-binding RPs in response to nucleolar stress. Also, it is unknown whether RPL11 forms an MDM2-associated subribosomal complex with a small group of RPs in response to nucleolar stress in vivo. Moreover, how many more RPs will be identified in this subribosomal complex that forms in response to nucleolar stress? The number of newly reported MDM2-binding RPs has continued to increase, even though some RPs such as RPL29, RPL30, and RPS19 have been excluded from this list.42,131 The last and most challenging question is, what is the biochemical mechanism governing the inhibition of MDM2 by these RPs? Although previous and recent studies55,103 performed by our group suggested that the binding of RPs to MDM2 may cause a conformational change in MDM2 that alters its tertiary structure within its central region, this change might reduce its binding affinity for p53, thus weakening its ability to ubiquitinate p53. Completely solving this puzzle will require more careful and systematic biochemical and biophysical studies. Solving the crystal structure of the MDM2-RP-p53 complex in the near future will accelerate progress in this field.
New insights into the molecular and structural basis of the RP-p53 pathway will also provide more opportunities for anticancer drug development. It is now recognized that targeting both MDM2 and MDMX would be a more effective way to activate p53 and suppress tumor growth. 132 As a matter of fact, the Hsp90 inhibitor, 17AAG, antagonizes MDMX and works synergistically with Nutlin-3 to disrupt the association of MDM2 with p53 and activate p53-dependent apoptosis in solid tumors. 133 An ideal anticancer drug candidate would be a small molecule that could bind to the zinc finger domain of MDM2 and prevent its inhibition of p53 in a manner similar to RPL11.52,53,103 This hypothetical molecule could potentially activate p53 by deactivating both MDM2 and MDMX, as turning on the nucleolar stress-RP-p53 pathway can simultaneously disarm both MDM2 and MDMX, as described earlier on in this essay. Therefore, designing lead compounds that mimic RPL11’s ability to bind to MDM2 would open a door for the future development of anticancer therapeutics.
Footnotes
Acknowledgements
We thank Emily Carron for proofreading.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by NIH-NCI grants CA127724, CA095441, and CA129828 to H.L.