
Abstract
The contribution of Mdm2, and its recently identified family member Mdmx (Mdm4), to tumorigenesis has primarily focused on their negative regulation of the p53 tumor suppressor. Although Mdm2 and Mdmx clearly inhibit p53, which can lead to tumor development, both have also been shown to affect tumorigenesis independent of p53. Given that Mdm2 and/or Mdmx overexpression is common and likely underestimated in human cancers, understanding the functions of these proteins beyond p53 control is critical. In recent years, new functions of Mdm2 and Mdmx that lead to genome instability, a hallmark of malignancy, have emerged. Specifically, roles in the DNA damage response that are distinct from their regulation of p53 have been identified. Inhibition of p53 as well as other components of the DNA damage response by Mdm2 and Mdmx can result in delayed DNA repair and increased genome instability, making Mdm2 and Mdmx a danger to the genome when aberrantly expressed. However, the genome instability caused by altered levels of Mdm2 and Mdmx could be used therapeutically for the treatment of cancer. Specifically, drugs/small molecules that target the interaction between Mdm2 and p53 can stabilize Mdm2, resulting in negative consequences on the genome that could be exploited for cancer treatment, particularly malignancies lacking functional p53.
Genome Instability and DNA Damage Contribute to Cancer
Genome instability, also commonly referred to as chromosome instability, is an established hallmark of cancer and postulated to contribute to the development and progression of malignancies. 1 Genome instability can manifest as structural and/or numerical chromosome aberrations, such as chromosome and chromatid breaks, insertions, deletions, translocations, amplifications, aneuploidy, and polyploidy.2,3 Chromosome aberrations are linked to tumorigenesis, as translocations leading to the activation of oncogenes, such as Myc, 4 or the deletion/mutation and subsequent loss of function of tumor suppressors, such as p53 or Rb,5,6 lead to cancer development. Cells that acquire the necessary genomic alterations that result in uncontrolled proliferation and evasion of apoptosis are disposed to becoming a cancer cell.1,5
Although the causes of genome instability are incompletely understood, data show DNA damage and the inability to properly repair this damage contribute to genome instability.7,8 DNA damage occurs from factors both internal and external to the cell, and consequently, cells have evolved sophisticated DNA damage signaling and repair pathways to maintain genome integrity. An immediate DNA damage response is activated upon genotoxic insult that engages many proteins, including p53, the Mre11-Rad50-Nbs1 DNA repair complex, and DNA damage kinases ATM, ATR, Chk1, and Chk2, to activate cell cycle checkpoints, mark the damaged DNA, signal for DNA repair proteins, and ultimately repair the DNA damage.7-9 Alterations in DNA damage signaling or in the timing or fidelity of DNA repair can result in mild to severe chromosome abnormalities. 8 For example, DNA breaks can serve as substrates for translocations and fusions, which occur when breaks are not repaired correctly.2,10 Cells that have irreparable DNA damage or chromosome aberrations incompatible with life undergo apoptosis, which can be mediated by p53.11-13 If a cell acquires the ability to bypass the DNA damage cell cycle checkpoints and apoptosis, such as by inactivating the p53 pathway, this can lead to transformation and tumorigenesis. Individuals born with mutations in p53 or other genes in the DNA damage response have increased genome instability and incidence of cancer. For example, humans born with mutations in p53 or the DNA damage activated kinase CHK2 develop Li-Fraumeni or Li-Fraumeni–like syndrome and have a greatly increased rate of cancer development.13-15 Moreover, people with mutations in any of the 3 components of the Mre11-Rad50-Nbs1 DNA repair complex or the DNA damage kinase ATM have delays in DNA repair, which lead to increases in chromosomal abnormalities, genome instability, radiation sensitivity, and cancer incidence. 8 Therefore, proteins capable of inhibiting p53 and other proteins in the DNA damage response, resulting in loss of DNA damage-induced cell cycle checkpoints and apoptosis and increased genome instability, would provide a significant advantage to cells during tumorigenesis, and likely be harnessed by cancer cells to ensure their progression. Mdm2 and Mdmx may be such proteins (Fig. 1).
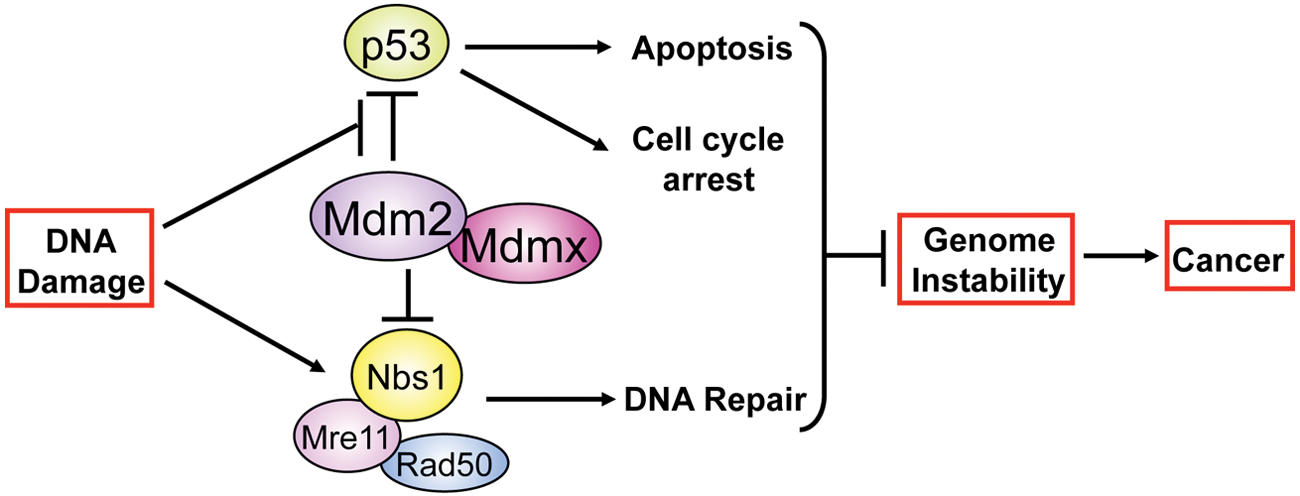
Mdm2 and Mdmx function in the DNA damage response. DNA damage results in the activation of p53 and the Mre11-Rad50-Nbs1 DNA repair complex. The DNA damage response relieves the inhibition of p53 by Mdm2/Mdmx, allowing p53 to induce cell cycle arrest or apoptosis. Cell cycle arrest gives cells time to repair DNA, but if DNA damage is severe or cannot be repaired, apoptosis ensues. The Mre11-Rad50-Nbs1 complex has essential roles in the DNA damage response and repair of DNA, and Mdm2/Mdmx binding to Nbs1 modulates these. Consequently, when Mdm2 or Mdmx is overexpressed, DNA repair is significantly delayed, causing damage to remain for longer, and p53 is also inhibited, resulting in a reduction in cell cycle arrest and apoptosis. These promote genome instability and tumorigenesis. Therefore, Mdm2 and Mdmx are particularly detrimental to the genome when expressed at elevated levels.
Mdm2, Mdmx, and p53 in Cancer
p53 is regulated by Mdm2 and Mdmx individually, but their heterodimerization cooperates to effectively block p53 transcriptional activity (Figs. 1 and 2A).16,17 Since deletion of p53 rescues the lethality of Mdm2 or Mdmx deletion during murine embryogenesis,18-20 many assume the sole function of Mdm2 and Mdmx is to regulate p53. However, these results only highlight the fact that Mdm2 and Mdmx are required to regulate p53 during embryogenesis, a time of tremendous proliferation when p53 would need to be held in check, and do not rule out other functions with different proteins. Moreover, Mdm2 and Mdmx are overexpressed in many malignancies,21-23 but it is unclear whether increased levels of Mdm2 and Mdmx affect the same proteins as normal levels. While many human cancers may select for Mdm2 and/or Mdmx overexpression as a mechanism of p53 inhibition, tumors that have mutated or deleted p53 can also express elevated levels of Mdm2 and/or Mdmx.24-29 Thus, Mdm2 and Mdmx may have functions independent of p53 that are selected for during tumorigenesis. Furthermore, it is likely that the frequency of increased levels of Mdm2 in human malignancies is grossly underestimated due to technical issues with Mdm2-specific antibodies and a lack of scoring 2- to 4-fold increased protein as elevated. Because of the identification of SNP309 in the promoter of MDM2, it is now appreciated that 2- to 4-fold increased levels of Mdm2 enhance cancer susceptibility in specific patient populations. 30 A mouse model of this polymorphism supports this finding. 31 Prior to the SNP309 discovery, 4-fold elevated expression of Mdm2 in Mdm2 transgenic mice was shown to increase tumor formation. Notably, in the absence of p53, Mdm2 transgenic mice developed an altered tumor spectrum compared with p53-null mice, suggesting p53-independent effects of Mdm2. 32 As for Mdmx, two individual Mdmx transgenic mouse lines where Mdmx expression was driven by the chicken β-actin promoter spontaneously developed malignancies, 33 whereas another mouse model using an artificial promoter in the Rosa26 locus to express Mdmx did not. 34 The differences in cancer susceptibility among the Mdmx transgenic lines are postulated to be due to differences in the levels of Mdmx expression, but this will need to be tested. The in vivo data could also suggest that the role of Mdmx in tumorigenesis is more complicated than what is known for Mdm2. These and other results indicate Mdm2 and Mdmx overexpression contribute to tumor development and that there is likely a p53-independent component.
Mdm2 in Genome Instability
Given that Mdm2 inhibits p53, it would be reasonable to assume that cells with wild-type p53 should have a growth/survival advantage when Mdm2 is overexpressed. However, investigators in the Mdm2/p53 field have repeatedly attempted to generate stable cell lines that contain wild-type p53 and grossly overexpress Mdm2 but have been unsuccessful.35-37 High levels of Mdm2 are also not tolerated in cells in vivo.32,34 However, cell lines with mutated or deleted p53 can survive prolonged Mdm2 overexpression. At an international Mdm2 meeting several years ago, Dr. Carol Prives questioned why Mdm2 had this effect and challenged those in the field to solve this enigma. Investigations in recent years have revealed novel Mdm2 functions that have begun to provide an explanation. Although there are data to suggest Mdm2 overexpression induces a p53-independent cell cycle arrest, 38 which would block proliferation, others have shown Mdm2 can target for proteosomal degradation Rb,39,40 the cell cycle inhibitor p21,41,42 and Foxo3a,43,44 a transcriptional regulator of cell cycle inhibitors, which would increase proliferation. Importantly, Mdm2 overexpression is reported to induce genome instability. Specifically, Mdm2 overexpression (2- to 4-fold) in cultured fibroblasts expressing wild-type p53 results in centrosome hyperamplification and chromosome instability. 37 Elevated levels of Mdm2 (4-fold) in Mdm2 transgenic mice increase chromosome and chromatid breaks, chromosome fusions, aneuploidy, and polyploidy in vivo prior to the development of cancer with the incidence of these chromosomal alterations increasing with age.45,46 Notably, when levels of Mdm2 are decreased, as in Mdm2 heterozygous primary murine fibroblasts that contain wild-type p53, there are decreased chromosome aberrations and increased genome stability. 47 Although these studies did not evaluate the role of Mdm2-mediated p53 inhibition versus a p53-independent function of Mdm2 in the genome instability observed, altering Mdm2 levels appears to have significant consequences on chromosome stability. In addition, the data suggest cells that overexpress Mdm2 and have functional p53 are still able to sufficiently activate p53, preventing cell proliferation or survival of genomically unstable cells. Therefore, only a rare cell that has acquired the genetic alterations necessary to sustain prolonged elevated levels of Mdm2 and increased genomic instability could develop into a cancer. This is exemplified in Mdm2 transgenic mice, which have evidence of significant genome instability as young as 5 months old, but do not develop cancer until later in life and live significantly longer than p53-null mice.32,46,48
Evidence that Mdm2 affects genome stability independent of its interactions with p53 has also emerged. Increased polyploidy was observed in vivo in mammary epithelial cells with elevated Mdm2 levels, and remarkably, this was also evident in Mdm2 overexpressing mammary epithelial cells lacking p53. 49 It is conceivable, but not yet tested, that Mdm2 targeting Rb for proteosomal degradation contributes to genome instability, since a deficiency in Rb has been recently linked to chromosome instability.6,39,40 However, the polyploidy in Mdm2 overexpressing mammary epithelial cells was not rescued with loss of E2F1, a transcription factor regulated by Rb. 50 Consequently, if reduced levels of Rb are involved in the genome instability caused by Mdm2 overexpression, it appears independent from the activation of E2F1, and therefore, either other E2F family members would contribute to the phenotype or the E2F-independent function of Rb that regulates chromatin structure would be responsible. 6 Notably, through a nonbiased screen for novel protein interactors of Mdm2, the Mre11-Rad50-Nbs1 DNA repair complex was identified. 51 Mdm2 overexpression was shown to inhibit DNA double-strand break repair mediated through a novel, direct interaction between Mdm2 and Nbs1 and independent of p53 (Fig. 1).51,52 Regardless of p53 status, increased levels of Mdm2, but not Mdm2 lacking its Nbs1-binding domain, caused delays in DNA break repair, chromosomal abnormalities, and genome instability. 52 These data demonstrated Mdm2-induced genome instability can be mediated through Mdm2:Nbs1 interactions and independent from its association with p53. In addition, it was determined that the small molecule Mdm2:p53 binding inhibitor, Nutlin-3a, stabilized Mdm2, leading to increased levels in cells. 53 Pharmacological stabilization of Mdm2 induced DNA double-strand breaks and a DNA damage response, and this could occur in the absence of p53.54,55 Therefore, although inhibition of p53 by Mdm2 may lead to some forms of genome instability, evidence is mounting that Mdm2 has functions that contribute to genome instability independent of its regulation of p53.
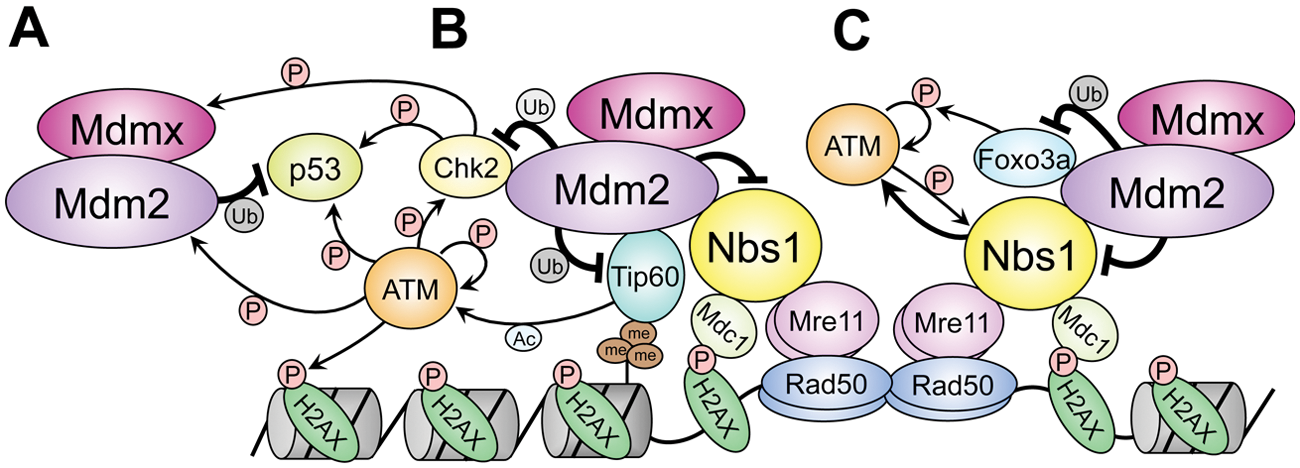
Mdm2 interacts with and inhibits multiple proteins in the DNA damage response. (
Mdmx and Chromosome Instability
Mdmx overexpression due to amplification or an undefined mechanism has been observed in many human cancers.22,23 Due to the negative regulation of p53, overexpression of Mdmx is expected to alter genome integrity at least somewhat. However, p53-independent effects of Mdmx on genome stability have been recently revealed. In contrast to Mdm2, loss of Mdmx is associated with genome instability. Specifically, in Mdmx/p53-double null mice, the development of spontaneous tumors occurred earlier than in p53-null mice. Murine embryonic fibroblasts from Mdmx/p53-double null mice exhibited multipolar mitosis and subsequent loss of chromosomes. These genomic alterations were not observed in Mdm2/p53-double null cells, indicating the loss of Mdmx promotes genome instability in a nonredundant manner from Mdm2.56,57 Additionally, in recent studies, we have observed that elevated levels of Mdmx lead to inhibition of DNA double-strand break repair, increased chromosomal alterations, and in vitro transformation independent of p53 (Melo and Eischen, manuscript in preparation). High levels of Mdmx were shown to be lethal in developing embryos, and although genome instability was not investigated, this could not be rescued with loss of p53. 34 Together the data suggest a p53-independent function by which altered Mdmx levels (decreased or increased) induce genome instability, which likely contributes to tumorigenesis. This differs from Mdm2, which induces genome instability when overexpressed and increases genome stability when expressed at reduced levels.37,45-47,49,51,52 Since relatively little is known about Mdmx, it will be important to determine the mechanism of Mdmx function in genome instability to define differences from Mdm2 function or whether its effects on the genome are due to its interactions with Mdm2.
Mdm2 and Mdmx in the DNA Damage Response
DNA damage induces complex signaling cascades that include many proteins, the identity of which depend on the type and extent of the DNA damage. For example, DNA double-strand breaks are sensed by the Mre11-Rad50-Nbs1 complex, which catalyzes the activation of the DNA damage-induced kinase ATM. 8 ATM then phosphorylates hundreds of proteins involved in the DNA damage response, including Nbs1, Mdm2, Mdmx, p53, histone H2AX, and others (Fig. 2B,C). 58 Currently, the function and consequence of the multitude of phosphorylation events that occur during the DNA damage response are incompletely understood. However, phosphorylation can alter protein:protein interactions, activate kinases and other enzymes, change protein stability, and alter cellular localization, among other consequences. For example, following DNA damage, phosphorylation of Mdm2, Mdmx, and p53 induces Mdm2:Mdmx interaction, inhibits Mdm2:p53 association, and may inhibit or promote Mdmx:p53 binding. 17 DNA damage also leads to an increase in the E3 ubiquitin ligase activity of Mdm2 that appears to be regulated by phosphorylation and is reported to result in the degradation of both itself and Mdmx. 17 However, due to commonly used antibodies that do not recognize phosphorylated Mdm2, the destabilization of Mdm2 following DNA damage is now controversial.59-65 Mdmx can, paradoxically, facilitate Mdm2-mediated p53 ubiquitination, but it is also reported to stabilize p53; this is postulated to be due to the relative levels of Mdmx and Mdm2 in the cell.16,17 Ultimately, posttranslational modifications of Mdm2, Mdmx, and p53 are thought to allow p53 activation following DNA damage.17,66-68 Additionally, DNA damage-induced phosphorylation events and other posttranslational modifications also likely promote alterations in protein interactions between Mdm2 and Mdmx and other proteins involved in the DNA damage response. For example, Mdm2 is reported to bind and facilitate Chk2 kinase ubiquitination and degradation independent of its own ubiquitin ligase activity (Fig. 2B). Phosphorylation of Chk2 by ATM following DNA damage decreases its ability to bind Mdm2, presumably allowing Chk2 to function in the DNA damage response. 69 Chk2 phosphorylates Mdmx and p53; the phosphorylation of p53 is necessary for its activation following DNA damage.68,70 It will be necessary to test whether higher levels of Mdm2 in cancer cells bind more Chk2, albeit weakly, leading to increased degradation of Chk2, suppression of the DNA damage signal, and a delay in the activation of p53.
There are a number of studies that link Mdm2 to the DNA damage response independent from its role as a regulator of p53. Mdm2 has been reported to associate with, as well as alter the function or stability of, proteins other than p53 in the DNA damage response, such as ATM, Nbs1, Tip60, Foxo3a, Mdc1, Kap1, and PolH.43,44,51,60,71-74 Specifically, Mdm2 was shown to co-immunoprecipitate with ATM, but it is unresolved as to whether the interaction between the two is direct or mediated through another protein both bind, such as Nbs1 or the histone acetyltransferase Tip60.51,60,71 Activation of ATM is catalyzed by the Mre11-Rad50-Nbs1 complex and acetylation by Tip60 (Fig. 2B).8,75 We have shown Mdm2 directly binds Nbs1 of the Mre11-Rad50-Nbs1 complex and inhibits DNA double-strand break repair independent of p53 (Figs. 1 and 2). 51 Unexpectedly, this function of Mdm2 did not require its E3 ubiquitin ligase activity or its RING domain.51,52 Only the region of Mdm2 that interacted with Nbs1 was necessary and sufficient to delay DNA break repair. Moreover, the Mdm2 binding domain of Nbs1 was also required for Mdm2 to inhibit DNA repair. The mechanism for this novel function of Mdm2 appears to be inhibition of the early DNA damage response signal mediated by ATM, as Mdm2 overexpression results in a delay in phosphorylation of histone H2AX, an ATM target and mark of DNA breaks that occurs early in DNA repair. Data also show a delay in the appearance of phosphorylated ATM/ATM target protein foci at sites of DNA damage, and this required the Nbs1 binding domain of Mdm2. 52 Thus, Mdm2 inhibition of DNA break repair is independent of its E3 ubiquitin ligase activity but dependent on its interaction with Nbs1. Recently, Mdm2 was also shown to bind and polyubiquitinate Tip60 and Foxo3a, targeting them for degradation (Fig. 2B,C).43,44,71 Loss of Tip60 function results in unrepaired DNA breaks, a consequence of reduced ATM activation.75,76 Foxo3a transcriptionally controls genes important for cell cycle arrest, but also genes in DNA repair. 77 Foxo3a also has been shown to bind and facilitate the autophosphorylation of ATM, which is necessary for its activation (Fig. 2C). Cells lacking Foxo3a have an impaired ATM-mediated DNA damage response following gamma irradiation and an inhibition of DNA break repair. 78 Currently, it remains to be determined whether Mdm2:Tip60 and Mdm2:Foxo3a interactions lead to reduced ATM activation and DNA repair, but the framework for these possibilities exists. Therefore, increased levels of Mdm2 lead to the inhibition of Nbs1 and the degradation of Tip60 and Foxo3a, each of which would result in the suppression of ATM activation and a delay in DNA repair; these are perfect conditions for the development of genome instability.
Less is known about the interaction of Mdm2 with Kap1 and DNA polymerase eta (PolH), both of which could affect genome stability. Mdm2 binds Kap1, a heterochromatin protein that is phosphorylated by ATM following DNA damage. 72 Phosphorylated Kap1 is thought to promote the opening (decondensation) of chromatin to allow access to DNA repair proteins. 79 Kap1 was shown to facilitate Mdm2-mediated p53 ubiquitination, 72 but the effects of Mdm2:Kap1 binding on DNA repair itself have not been investigated. Mdm2 was also recently shown to bind and polyubiquitinate PolH, an enzyme involved in nucleotide excision repair, following UV irradiation, resulting in its proteosomal degradation. 74 Humans with defects in PolH develop xeroderma pigmentosum variant syndrome (XPV), are sensitive to UV irradiation, and have increased incidence of skin cancer. 80 Since the experiments on Mdm2 and PolH were performed in cell lines, it may be worthwhile testing the role of this interaction in mice to determine whether it confers increased sensitivity to UV radiation. These data also suggest that Mdm2 may regulate multiple DNA repair pathways, since UV irradiation causes a different kind of DNA damage than gamma irradiation.
In addition to altering protein interactions, DNA damage changes the cellular localization of both Mdm2 and Mdmx. Since Mdm2 and Nbs1 directly bind, 51 and Nbs1 is known to recruit multiple proteins to sites of DNA damage, 8 it is likely that Mdm2 localizes to DNA breaks through its interaction with Nbs1. However, Mdm2 was also reported to associate with and be stabilized by Mdc1, a scaffold protein that mediates the interaction between Nbs1 and phosphorylated H2AX to retain Nbs1 at sites of DNA damage (Fig. 2).73,81 Although not proven, it is likely the interaction of Mdm2 with Mdc1 is mediated through Nbs1. Mdm2 also binds Tip60, but Tip60 is recruited to DNA breaks by Nbs1,71,75 so again Nbs1 is expected to be the key to localizing Mdm2 to DNA damage. Regardless of how Mdm2 moves to DNA damage, its association with proteins that go to sites of DNA breaks should result in increased concentrations of Mdm2 at chromatin, allowing it to inhibit DNA repair. In support of this, Mdm2, a primarily nuclear protein, normally appears diffusely nuclear by immunofluorescence but, following gamma irradiation, shows a punctate staining pattern that partially co-localizes to sites of DNA damage and with the Mre11-Rad50-Nbs1 complex. 51 Moreover, recent biochemical data show increased levels of Mdm2 at chromatin and bound to Nbs1 after gamma irradiation (Melo and Eischen, manuscript in preparation). Therefore, the DNA damage response proteins that associate with Mdm2 and Nbs1 may interact together at sites of DNA damage (Fig. 2). As for Mdmx, it primarily resides in the cytoplasm and moves into the nucleus after DNA damage. Phosphorylation of Mdmx promotes its nuclear localization and Mdm2-mediated ubiquitination, leading to its degradation.17,82-85 Although it has been shown to participate, Mdm2 is not required for the nuclear localization of Mdmx, as it can move to the nucleus in cells lacking Mdm2.82,85 The consequences of Mdmx nuclear localization remain unresolved. Why would a cell spend the energy to shuttle Mdmx into the nucleus just to be degraded? Instead, Mdmx was recently shown to bind p53 and facilitate its binding to the Mdm2 promoter following DNA damage. 86 Although Mdmx may function with or without Mdm2 to regulate p53, it is also likely regulating other proteins independent of p53 and possibly independent of Mdm2 in the nucleus. For example, we have detected Mdmx at chromatin following DNA damage and in association with Nbs1 independent of p53 (Melo and Eischen, manuscript in preparation). Dr. David Lane has also observed Mdmx:Nbs1 association using a quantitative proteomic approach (Cindy Coffill and David Lane, personal communication). Therefore, Mdm2 and Mdmx localization after DNA damage, and the proteins with which they associate, provide important insight into their genomic functions and reveal further links to the DNA repair process. With the data generated thus far, a model is emerging that indicates Mdm2 and likely Mdmx function in the early DNA damage response and influence not only p53 but other proteins that are critical for DNA repair and maintenance of the genome (Fig. 2).
A question arises as to whether only increased levels of Mdm2 and Mdmx inhibit DNA break repair or whether they have this function under normal physiological conditions. Data exist for Mdm2 that provide insight into this question. Notably, murine fibroblasts expressing a mutant of Nbs1 that could not interact with endogenous Mdm2 repaired DNA breaks at a faster rate than cells expressing wild-type Nbs1. 52 These results suggest that physiological levels of Mdm2 normally slow Nbs1-mediated DNA repair. This intriguing observation then begs the question of why a cell would need to slow DNA repair. It is possible that DNA repair, which is an orchestra of hundreds of dynamic proteins and a process that needs to be done perfectly, requires a specific rate to ensure repair is completed with high fidelity. Mdm2 appears to fine-tune this process, but when it is overexpressed, it decelerates DNA repair too much, which leads to chromosome abnormalities and genome instability. It will be important in the future to determine the influence Mdmx has on this function of Mdm2.
Targeting Mdm2 and Mdmx in Cancer
Pharmacologically targeting Mdm2 and Mdmx binding to p53 has been a recent focus in cancer therapeutics with the intention of releasing p53 from Mdm2 and Mdmx, leading to tumor cell apoptosis or cell cycle arrest. While proof of principle has been achieved in vitro and in mouse models, it has not yet translated into clinical success. 87 In addition, although these inhibitors hold great promise for treating some cancers, constitutive activation of p53 by oncogenes, decreased levels of Mdm2, or an Mdm2 inhibitor selects for tumors that have inactivated p53.88-91 Moreover, half of human cancers lack functional p53 at the time of diagnosis. 13 However, with recent studies highlighting p53-independent functions of Mdm2 and Mdmx in genome instability, therapy designed to capitalize on this function of Mdm2 and Mdmx would have a broader cancer therapeutic application and may be more efficacious in malignancies that have inactivated p53, as these cancers have lost checkpoint control. The genome instability induced by Mdm2 and Mdmx overexpression should lead to novel, possibly synthetic lethal, approaches to cancer treatment. For example, gamma irradiation of sarcoma cells overexpressing Mdm2 showed a decrease in colony formation. 92 This suggests combined therapies that use an Mdm2 inhibitor that results in increased Mdm2 levels (e.g., Nutlin-3a or MI-63) in combination with radiation or chemotherapeutics that induce DNA damage may be an effective targeted therapy even in cancers that have lost functional p53; several studies support this concept. 87 For example, Nutlin-3a radiosensitized p53 mutant or p53-null prostate cancer cells. 93 Nutlin-3a also increased the cell death of sarcoma and cutaneous T cell lymphoma cells with or without functional p53 in the presence of doxorubicin or cisplatin, which causes DNA damage.94,95 Recently, it was shown that gemcitabine, which causes DNA replication stress and subsequent breaks, together with MI-63 showed cooperativity and increased tumor cell death. 96 These and other data suggest a therapeutic benefit of combining DNA damaging agents with small molecules/compounds that increase the levels of Mdm2 to kill cancer cells that express or lack functional p53; a similar approach could be taken with Mdmx.
Conclusions
The accumulating data point increasingly to other functions for Mdm2 and Mdmx in the DNA damage response, in addition to their role in regulating p53 (Figs. 1 and 2). Results reveal that elevated levels of Mdm2 or Mdmx, as observed in human malignancies, have detrimental effects not only on p53 but also on other proteins in the DNA damage response. The consequence of this is genome instability, which would facilitate the acquisition of genetic alterations that could induce cellular transformation and/or permit a cancer cell to survive in harsh (low nutrient, low oxygen) environments, evolve into a more aggressive metastatic malignancy, and possibly develop resistance to certain treatments. However, too much genome instability is detrimental and leads to cell death, even of cancer cells. As a result, inducing DNA damage or additional genome instability could be used as an avenue for targeted therapy of the many cancers that overexpress Mdm2 and/or Mdmx. Moreover, combination therapies that increase Mdm2 and/or Mdmx levels concomitant with DNA damage could be effective to specifically target the 50% of malignancies that have inactivated p53 and have therefore, lost DNA damage checkpoint control. As our understanding of Mdm2 and Mdmx improves, it expands to include new functions that likely contribute to tumorigenesis and thus warrant further investigation. Importantly, these novel functions of Mdm2 and Mdmx should be able to be exploited for cancer treatment.
Footnotes
Acknowledgements
We thank the members of the Eischen lab for helpful discussions and critical review of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from NIH/NCI: ANM is supported by F31CA150546, and CME is supported by R01CA117935, R01CA160432, and the Cancer Center grant P30CA068485.