
Abstract
Mdm2 is an essential regulator of the p53 tumor suppressor. Mdm2 is modified at transcriptional, post-transcriptional, and post-translational levels to control p53 activity in normal versus stressed cells. Importantly, errors in these regulatory mechanisms can result in aberrant Mdm2 expression and failure to initiate programmed cell death in response to DNA damage. Such errors can have severe consequences as evidenced by tumor phenotypes resulting from amplification at the Mdm2 locus and changes in post-transcriptional and post-translational regulation of Mdm2. Although Mdm2 mediated inhibition of p53 is well characterized, Mdm2 interacts with many additional proteins and also targets many of these for proteosomal degradation. Mdm2 also has E3-ligase independent functions and p53-independent functions that have important implications for genome stability and cancer.
Introduction
Murine double minute 2 (Mdm2) is a RING finger containing E3 ubiquitin ligase that negatively regulates p53 by inhibiting p53-mediated transactivation of target genes and targeting it p53 for proteosomal degradation (reviewed in Marine et al. 2006).1 The critical nature of the p53-Mdm2 relationship was first observed in animal models. Specifically, loss of Mdm2 leads to embryonic lethality very early in development that is rescued by concomitant deletion of p53,2,3 and Mdm2 loss in all cell types examined results in a cell lethal phenotype.4-7 In addition, although Mdm2 heterozygosity is sufficient to dampen p53 levels under normal conditions in an adult mouse, a low dose of ionizing radiation is lethal to these mice in a p53-dependent manner. 8 The data suggest that a 50% gene dose of Mdm2 is sufficient to maintain appropriate p53 levels except in response to DNA damage. Mice carrying a hypomorphic and a null allele of Mdm2, which express only 30% of wild-type Mdm2 levels, and are small, lymphopenic, and radiosensitive, have even greater p53 activity. 9 The p53-dependency of all these phenotypes indicates that Mdm2 primarily functions to inhibit p53 activity in development and in response to DNA damage.
The mouse models described above demonstrate that Mdm2 and p53 expression levels must be tightly regulated for normal embryonic development and homeostasis. Errors in this signaling network can lead to aberrant expression of effectors of the p53 pathway, resulting in tumorigenesis.10,11 For example, approximately 30% of all sarcomas bear MDM2 amplifications, 12 and abnormal post-translational regulation of MDM2 protein also contributes to human cancers, as suggested by overexpression of MDM2 protein without MDM2 amplification in B-cell chronic lymphocytic leukemias and non-Hodgkin’s lymphomas. 13 Interestingly, Mdm2 does not have to be drastically overexpressed to affect tumor phenotypes. An animal model that exhibits a 3- to 4-fold increase in Mdm2 levels is tumor prone. 14 Additionally, a single nucleotide polymorphism (SNP) in the MDM2 promoter that results in 2- to 4-fold higher Mdm2 protein expression results in an increased risk in spontaneous tumor formation,15,16 whereas a 50% reduction in Mdm2 gene dosage delayed tumor formation in tumor prone mouse models.8,17-19 Thus, minor differences in Mdm2 levels have a large impact on tumor phenotypes.
Taken together, these in vivo studies highlight the importance of regulating Mdm2 expression during embryogenesis and in adult tissues. Since Mdm2 must be differentially expressed in normal cells versus stressed cells, there are numerous mechanisms in place to regulate Mdm2 expression in a context dependent manner. This review focuses on proteins that function to maintain appropriate levels of Mdm2 through transcriptional, post-transcriptional, and post-translational regulation. We also discuss the E3-ligase dependent and independent functions of Mdm2 and conclude with some remarks on p53-independent functions of Mdm2.
Proteins That Interact with Mdm2
Yeast 2 hybrid screens, mass spectrometry, and immunoprecipitation experiments have identified a large number of proteins that physically interact with Mdm2. These proteins are summarized in Tables 1 and 2 and are divided into 2 groups: proteins that function upstream of Mdm2 (effectors) to regulate Mdm2 at the transcriptional, post-transcriptional, or post-translation level (Table 1) and proteins that are specific downstream targets of Mdm2 (Table 2). Not all interactions have been examined at physiological levels. However, since Mdm2 is overexpressed in human cancers, the overexpression studies presented in this review are relevant for the characterization of Mdm2 binding partners and targets during tumorigenesis.
Effectors That Interact with MDM2
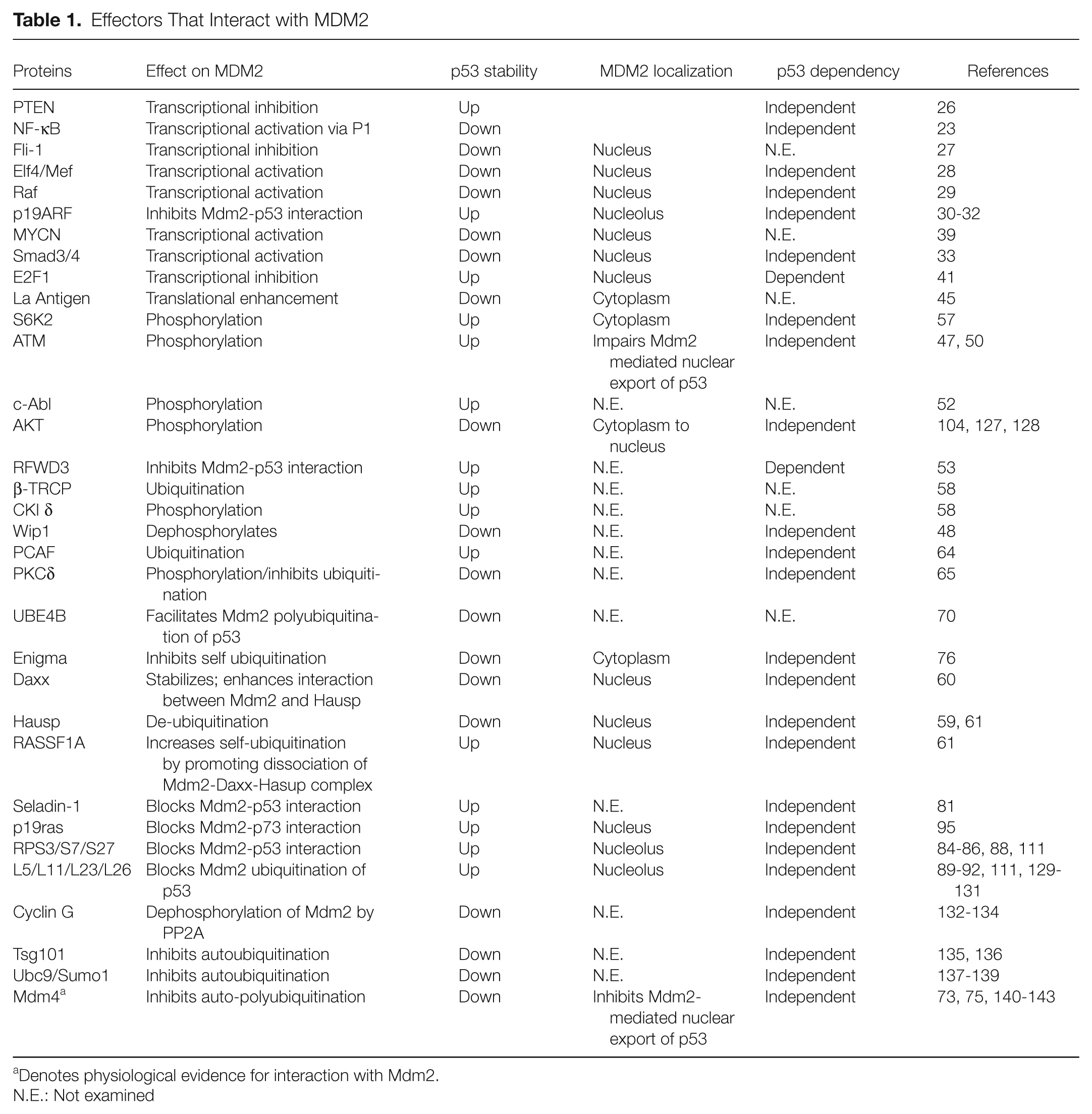
Denotes physiological evidence for interaction with Mdm2.
N.E.: Not examined
Downstream Targets of MDM2
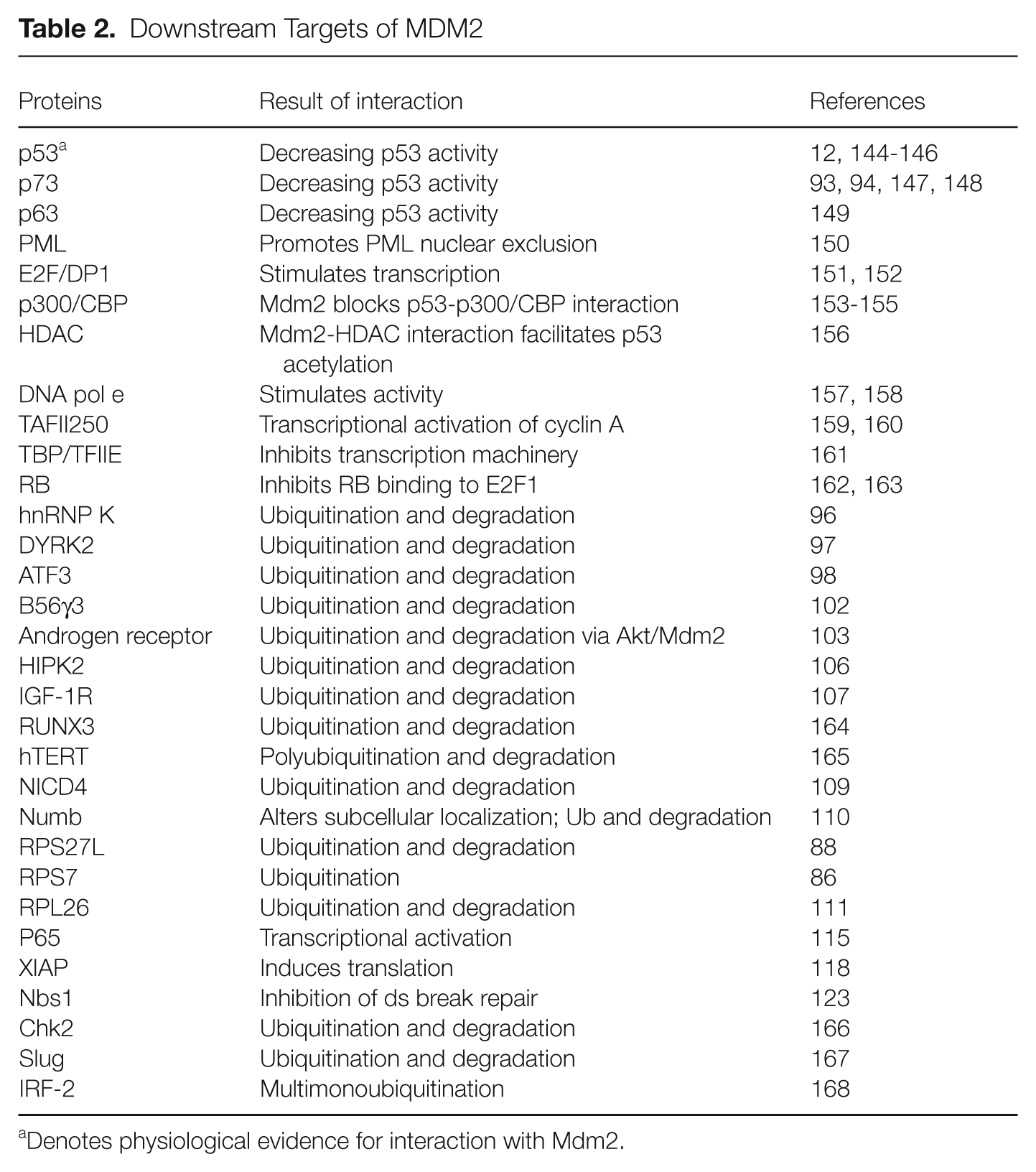
Denotes physiological evidence for interaction with Mdm2.
Effectors of Mdm2
The majority of proteins in this group regulate Mdm2 transcriptionally through direct binding at the P1 or P2 promoter, post-transcriptionally through binding to Mdm2 mRNA, or post-translationally through phosphorylation, ubiquitination, or de-ubiquitination of Mdm2.
Transcriptional regulation of Mdm2
Mdm2 transcription is regulated by 2 promoters named P1 and P2 that regulate basal and inducible expression, respectively.20-22 The P1 promoter is located upstream of the first exon, and the P2 promoter is located within the first intron (Fig. 1). Transcription from the upstream, constitutive promoter is p53-independent, 21 and to date there are few known regulators of the P1 promoter. However, a recent study identified an NF-κB site in the P1 promoter (Fig. 1). NF-κB is a transcription factor that regulates T lymphocyte cell activation and proliferation during an immune response. Chromatin immunoprecipitation (ChIP) in transfected Jurkat T cells demonstrates that the p50/RelA subunit binds to an NF-κB site referred to as κB2 in the P1 promoter, resulting in increased Mdm2 expression. Thus, survival of activated T cells is mediated in part through NF-κB induced Mdm2 expression through the P1 promoter. 23 The P1 promoter is also regulated, although not directly, by phosphatase and tensin homolog deleted on chromosome 10 (PTEN). PTEN is a tumor suppressor protein that negatively regulates the PI3K-Akt signaling pathway to suppress cell proliferation and induce apoptosis by blocking Mdm2-mediated p53 degradation.24,25 Reporter assays using transfected PTEN in the presence or absence of p53 demonstrate that PTEN negatively regulates the Mdm2 P1 promoter in a p53 independent manner, (Fig.1) but the transcription factor mediating this PTEN dependent regulation remains unknown. 26
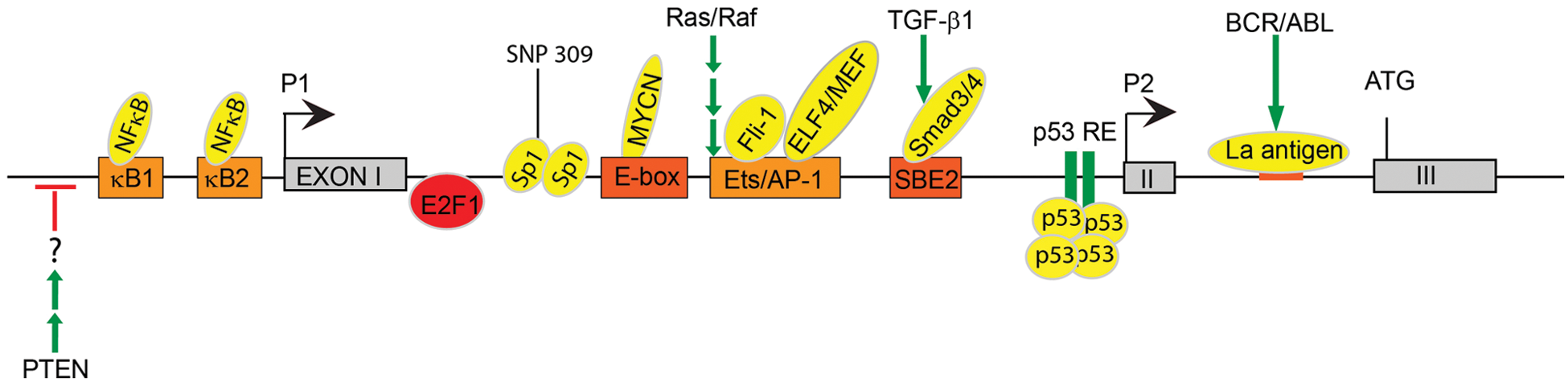
Transcriptional and post-transcriptional regulation of Mdm2. Mdm2 transcription is regulated by 2 promoters, P1 and P2. The P1 promoter is located upstream of the first exon and controls basal expression of Mdm2. The P2 promoter is located within the first intron and uses an alternate start site within the second exon. Since the P2 promoter is inducible, it functions to regulate Mdm2 expression after DNA damage. κB1 = NF-κB binding site 1 (TGAATTTCCT); κB2 = NF-κB binding site 2 (GGAAGTTTCC); E-box = Enhancer box; Ets/AP-1 = Ets/AP-1 binding element; SBE = Smad-binding element. Boldface denotes regulation that is p53-independent.
Unlike the P1 promoter, the inducible P2 promoter contains several known response elements for transcription factors that either inhibit or activate Mdm2 transcription depending upon the cellular conditions. For example, 2 members of the Ets family of transcription factors, Fli-1 27 and ELF4/MEF, 27 positively regulate Mdm2 expression at the transcriptional level through direct binding to the P2 promoter. Overexpression of Fli-1, combined with inactivation of p53, is implicated in the progression of Friend Murine Leukemia Virus. Analysis of tumors reveals a positive expression correlation between of Fli-1 and Mdm2 and a negative correlation between Fli-1 and p53, indicating that Fli-1 indirectly regulates p53 levels through its transcriptional regulation of Mdm2. 27 The Ets family member, ELF4/MEF, regulates cell cycle progression and has been implicated in tumorigenesis. ChIP analysis revealed that Elf4/Mef binds to and activates the Mdm2-P2 promoter. Mouse embryo fibroblasts (MEFs) isolated from Elf4/Mef null mice exhibit decreased Mdm2 expression and enhanced DNA damage-induced senescence coupled to an accumulation of p53 protein. In contrast, overexpression of Elf4/Mef results in increased Mdm2 expression and oncogenic transformation. Interestingly, Elf4/MEF/p53 null MEFs are transformation resistant, 28 indicating that Elf4/Mef may have p53-independent functions in tumorigenesis. ChIP analysis for Elf4/Mef binding at the P2 promoter must be performed in p53 null MEFs to determine whether Elf4/Mef regulation of Mdm2 is p53-independent.
Mdm2 transcription is also induced by activated Ras and Raf via Ets/AP-1 motifs in the Mdm2-P2 promoter. 29 Expression of a constitutively active Ras or Raf allele in p53 −/− MEFs results in induction of Mdm2 transcription through the P2 promoter, indicating that Ras/Raf induces Mdm2 expression in a p53-independent manner. After treatment with γ-radiation, activated Ras is able to abrogate the p53 stress response, which is consistent with the observed increased in expression. 29 However, upon DNA damage, this effect can be neutralized by expression of p19ARF, which physically interacts with and inhibits Mdm230-32 (discussed below in section covering post-translational regulation of Mdm2). Treatment with the DNA damaging reagent, adriamycin, in MEFs expressing p19ARF resulted in increased p53 expression compared with p19ARF-/- MEFs. 29 Taken together, these data indicate that the opposing effects of Ras/Raf induced p19ARF and Mdm2 function to regulate p53 expression in unstressed versus stressed cells.
TGF-β1 signaling also induces Mdm2 expression in a p53-independent manner. Recent work demonstrates that the mechanism behind this transcriptional regulation involves TGF-β1 activation of the Smad3/4 transcription factors. 33 Smad3 and Smad4 ChIP assays in cells treated with TGF-β1 reveal that Smad3/4 bind the Smad binding element 2 (SBE2) located at nucleotide –245 in the P2 promoter region of Mdm2 (Fig. 1). Smad binding to the promoter results in increased Mdm2 protein expression and subsequent destabilization of p53 in human cancer cell lines. Induction of Mdm2 is p53-independent as evidenced by an increase in Mdm2 expression after TGF-β1 treatment in 2 cell lines that lack functional p53 (transformed 293T cells and SKOV3 ovarian cancer cells). Interestingly, Smad3 activation corresponds with increased Mdm2 levels in murine mammary epithelial cells undergoing EMT in response to TGF-β1 treatment. Furthermore, analyses of late-stage human breast carcinomas revealed high levels of activated Smad3 and Mdm2. 33 These results are consistent with a role for TGF-β1/Smad-dependent activation of Mdm2 in late stage metastatic cancer and implicate p53-independent functions of Mdm2 in tumorigenesis.
The MYCN oncogene is a member of the MYC family of transcription factors and is amplified in a variety of tumors, including neuroblastomas. 34 MYCN induces both cell proliferation and apoptosis, which are opposing cellular processes. MYCN shortens the G1-S transition and increases cell proliferation35-37 while it simultaneously induces p53-mediated apoptosis (reviewed in van Noesel & Versteeg 38 ). MYCN functions as an oncogene when its pro-proliferative effects outweigh its pro-apoptotic effects, resulting in escape from p53-mediated apoptosis. A ChIP screen using an MYCN-amplified neuroblastoma cell line revealed that MYCN binds to a consensus E-box within the human MDM2-P2 promoter (Fig. 1). Accordingly, induction of MYCN resulted in increased endogenous MDM2 mRNA and MDM2 protein, and inhibition of MYCN resulted in decreased MDM2 expression. These data indicate that the MYCN oncogene inhibits p53 mediated apoptosis in neuroblastoma cells by direct binding to and activation of the MDM2 promoter. 39
Although most reported Mdm2 effectors positively regulate MDM2 transcription, MDM2 transcription can also be negatively regulated in a p53-dependent manner as demonstrated by the E2F family transcription factor, E2F1. E2F1 regulates cell-cycle progression and induces apoptosis in response to DNA damage (reviewed in Engelmann & Putzer 40 ). Overexpression of E2F1 in a human osteosarcoma cell line that expresses wild-type p53 (U2OS) leads to a reduction in MDM2 expression through inhibition of the MDM2 promoter. ChIP analysis in a wild-type and a p53-deficient cell line demonstrates that E2F1 binds the Mdm2-P2 promoter in a p53-dependent manner (Fig.1). Since DNA damage results in the up-regulation of both E2F1 and p53, Tian et al. 41 examined whether E2F1 inhibits p53 transactivation of the MDM2 promoter. They found that after UV irradiation, the promoter is drastically down-regulated in the presence of E2F1 compared with cells treated with E2F1 siRNA. 41 Taken together, these results indicate that E2F1 antagonizes p53 transactivation of MDM2 after DNA damage, and they reveal a mechanism for protecting p53 from degradation after DNA damage.
Last, Mdm2 itself is transcriptionally regulated by wild-type p53 via p53 response elements located in the Mdm2 P2 promoter (Fig.1).42-44 In response to DNA damage, Mdm2 is transcriptionally activated by p53, consequently blocking p53 function. This in turn results in less Mdm2 being transcribed, resulting in increased p53 activity. Thus, the levels of Mdm2 and p53 activity within a cell are regulated by this autoregulatory feedback loop. Deregulation of this loop by amplification of the Mdm2 locus or aberrant transcriptional or post-translational regulation of Mdm2 can have dire consequences for cell proliferation and results in tumorigenesis.
Given that Mdm2 expression must be tightly regulated to ensure proper p53 expression and activity, deregulation of any of these bindings protein can affect Mdm2 levels and potentially lead to tumorigenesis. For example, a T-to-G single nucleotide polymorphism (SNP) in the P2 promoter was identified in humans that increases the binding affinity of the Sp1 transcriptional activator for the Mdm2 promoter (Fig. 1). This leads to increased Mdm2 protein levels and, consequently, decreased protein stability. Importantly, the MDM2SNP309G allele is correlated with increased cancer risk in humans. To directly examine the effect of this SNP in tumorigenesis, Post et al. 16 generated 2 humanized Mdm2SNP309 murine alleles and found that the SNP309G allele results in increased Mdm2 levels (2- to 3.7-fold in spleens and thymi, respectively) and decreased p53 activity in response to IR. Importantly, this allele also results in increased cancer risk. 16 These data indicate that small increases in Mdm2 transcription can promote tumorigenesis and demonstrate that transcriptional regulation of Mdm2 is essential for proper Mdm2 expression. The generation of similar mouse models that interfere with binding of any of these proteins to the Mdm2 promoter will reveal the physiological impact of these interactions in vivo. It is possible that a transgenic mouse overexpressing an Mdm2-transcriptional inducer such as Smad3/4 or a knockout of a transcriptional inhibitor such as E2F1 may result in increased Mdm2 expression and tumorigenesis. Conversely, a knockout of a transcriptional inducer may reduce Mdm2 levels leading to p53-dependent embryonic lethality and/or increased radiosensitivity.
Post-transcriptional regulation of Mdm2
Mdm2 is also regulated at the post-transcriptional level by an RNA binding protein named La antigen. The BCR/ABL oncoproteins are responsible for the transformation of the hematopoietic stem cells resulting in chronic myelogenous leukemia (CML). Recent data show that BCR/ABL upregulates Mdm2 expression by enhancing mdm2 mRNA translation via La antigen, which recognizes a 27 nucleotide segment in the 5′ untranslated region of mdm2 mRNA (Fig.1). 45 A recent study found that the JAK2(V617F) gain of function mutation also affects the p53 response to DNA damage through upregulation of La antigen. JAK2(V617F) is a gain of function mutation that promotes cytokine-independent growth of myeloid cells and accounts for a majority of myeloproliferative neoplasms (MPN). Expression of JAK2(V617F) in ex vivo cultured CD34(+) cells from MPN patients resulted in the accumulation of MDM2, resulting from a La antigen-dependent increase in MDM2 mRNA translation. These results reveal one mechanism behind the gain-of-function activity of JAK2(V617F), which ultimately results in the aberrant degradation of p53 after DNA damage. 46
Post-translational regulation of Mdm2
During the DNA damage response (DDR), p53 is stabilized through post-translational modification of Mdm2 (Fig. 2). For example, after DNA damage, Mdm2 is phosphorylated by ATM at serine 395 (Fig. 2). This phosphorylated Mdm2 species cannot efficiently promote translocation of p53 from the nucleus to the cytoplasm, 47 resulting in increased p53 stability. Wild-type p53-induced phosphatase (Wip1), which is a p53 target gene, can dephosphorylate this same Mdm2 residue. This results in stabilization of Mdm2 and subsequent ubiquitination and degradation of p53. 48 Thus, the phosphorylation status of Mdm2-S395 regulates the p53 response after DNA damage and implicates Wip1 as a potential oncogene. Interestingly, Wip1 is amplified and overexpressed in a number of human cancers (reviewed in Le Guezennec & Bulavin 49 ).
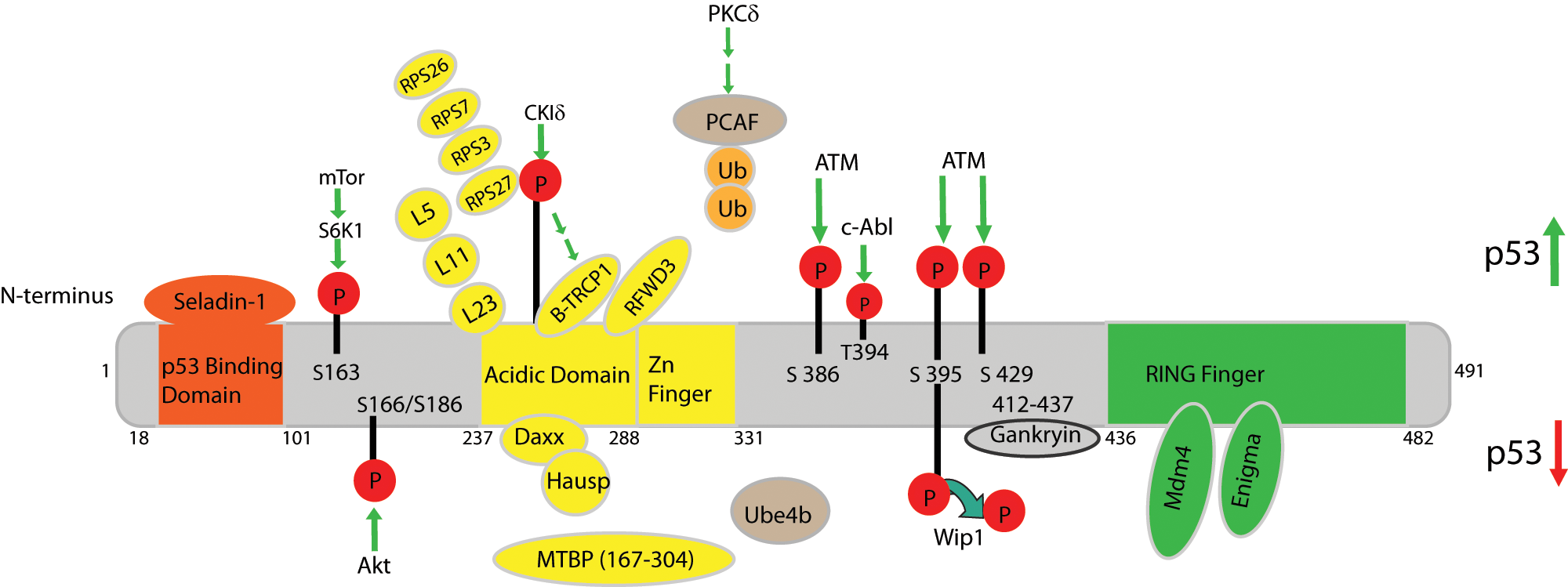
Effectors of MDM2. Schematic representing the characterized human MDM2 domains and known binding partners. Human MDM2 contains 491 amino acids and has 4 characterized domains: residues 18-101 comprise the p53 binding domain, residues 237-288 contain the acidic domain, which is adjacent to the Zinc finger domain, and the RING finger domain is within residues 436-482. MDM2 binding partners are depicted in orange, yellow, or green depending on the region of MDM2 to which they bind (orange = p53 binding domain, yellow = acidic domain/zinc finger, green = RING finger domain). Proteins whose binding locations on MDM2 are unknown are depicted in tan. Interactions that result in increased p53 stability and activity are depicted above the schematic, whereas interactions that result in decreased p53 stability and activity are shown below. P = phosphorylation; Ub = ubiquitination.
Other groups confirmed S395 as a direct substrate of ATM and revealed 2 additional phosphorylation sites (S386/429) adjacent to the RING domain (Fig. 2). Treatment with IR revealed that ATM phosphorylation of Mdm2 at these 2 sites prevents Mdm2-mediated polyubiquitination of p53 and is necessary for p53 stabilization. 50 ATM also activates the c-Abl kinase in response to DNA damage. 51 C-Abl in turn binds to and phosphorylates human MDM2 at Tyr 394, resulting in increased p53 activity. 52 In addition, the novel protein, E3 ligase RING finger and WD repeat domain 3 (RFWD3), which is another substrate of the checkpoint kinases ATM/ATR, was recently shown to interact with Mdm2 and p53, forming an RFWD3-Mdm2-p53 ternary complex. 53 Deletion analyses indicate that the Mdm2 acidic domain is required for its interaction with RFWD3. Consistent with its role as a negative regulator of Mdm2, overexpression of RFWD3 protects p53 from degradation in the presence of high Mdm2 levels after DNA damage. RFWD3 restricts polyubiquitination of p53 by Mdm2, resulting in increased p53 stability, thus inhibiting Mdm2-p53 interactions. 53 Together, these data indicate that ATM-mediated phosphorylation of Mdm2 is important for regulating the p53 response after DNA damage. Further studies using mouse models that disrupt ATM/c-Abl mediated phosphorylation and Wip1-mediated dephosphorylation of Mdm2 will be needed to determine the in vivo relevance of these modifications.
mTOR is an ATM/ATR-related protein kinase that functions to regulate cell proliferation, metabolism, and growth. 54 S6K1 is a downstream signaling molecule of mTOR 55 that promotes translation of various proteins through phosphorylation of the eukaryotic translation factor, eIF4B, and the ribosomal protein S6 (reviewed in Averous & Proud 56 ). A recent study shows that genotoxic stress caused by treatment with doxorubicin (which causes single and double stranded DNA breaks) results in phosphorylation of Mdm2 S163 by S6K1 in primary MEFs and 293 cells. Furthermore, immunoprecipitated S6K1 phosphorylated Mdm2 S163 in vitro (Fig. 2), indicating that Mdm2 S163 is a direct substrate of S6K1. Upon doxorubicin treatment, activated phosphorylated S6K1 tightly binds to Mdm2 (this interaction is independent of Mdm2 S163 phosphorylation), inhibits Mdm2 translocation to the nucleus, and prevents Mdm2-mediated p53 ubiquitination and degradation. 57 These data suggest that S6K1 indirectly regulates p53 stability through interacting with and phosphorylating Mdm2 after DNA damage. Although we do not know whether phosphorylation of S163 prevents binding of Mdm2 to p53, the end result of this phosphorylation is p53 stabilization. Therefore, these findings may have important implications for tumorigenesis and therapies that aim to retain Mdm2 in the cytoplasm and to stabilize p53.
Mdm2 stability is also regulated by the F-box ubiquitin ligase, β-TRCP, in a phosphorylation dependent manner. In response to DNA damage, Mdm2 interacts with β-TRCP1 and β-TRCP2 in vivo, resulting in p53 stabilization and apoptosis. In vitro phosphorylation assays demonstrate that CKIδ phosphorylates Mdm2 at multiple sites located within the acidic domain and in the N-terminal DSG site (Fig. 2). This phosphorylation is required for Mdm2-β-TRCP1 association and subsequent Mdm2 degradation. Interestingly, the E3-ligase activity of Mdm2 is dispensable for its degradation after DNA damage. However, co-expression of CKIδ and β-TRCP1 promotes degradation of an E3-ligase dead Mdm2 mutant as efficiently as wild-type Mdm2. These data indicate that CKIδ mediated phosphorylation rather than the RING domain is essential for regulating Mdm2 degradation after DNA damage. 58 Since Mdm2 is overexpressed in human tumors without amplification at the Mdm2 locus, it is possible that dysregulation of CKIδ and/or β-TRCP1 could result in increased Mdm2 protein stability and tumorigenesis.
HAUSP, a ubiquitin-specific protease, and the death domain-associated protein (Daxx) also regulate Mdm2 stability. Disruption of HAUSP in cell lines resulted in slow growth and failure to thrive as a consequence of p53 accumulation. However, this effect is not through Hausp-mediated de-ubiquitination of p53. Rather, Hausp positively regulates Mdm2 stability directly, resulting in p53 destabilization. 59 Mdm2 stability is also enhanced by Daxx, which physically interacts with Mdm2 in vitro and in vivo. In unstressed cells, co-immunoprecipitation assays and in vitro pull-down assays demonstrate that Daxx acts as a scaffold to physically connect Mdm2 to Hausp, preventing Mdm2 autoubiquitination, resulting in the stabilization of Mdm2. Upon DNA damage, Daxx dissociates from Mdm2, leading to Mdm2 autoubiquitination and subsequent degradation. 60 This dissociation is mediated at least in part by the tumor suppressor RASSF1A, which physically associates with Mdm2 and Daxx in the nucleus, disrupting the interactions between Mdm2, Daxx, and Hausp. 61 More recent studies demonstrate that Daxx itself is ubiquitinated by Mdm2 in vitro and in vivo. 62 Consistent with the in vitro data, loss of Daxx in mice leads to early embryonic lethality resulting from increased apoptosis, 63 indicating that Daxx functions to inhibit cell death during embryogenesis.
These data elucidate several mechanisms governing the cis- and trans-E3 activities of Mdm2 in stressed versus normal cells and demonstrate that Daxx plays a pivotal role in regulating Mdm2 self-ubiquitination.
P300-CBP-associated factor (PCAF), which is a histone acetyltransferase, exhibits intrinsic E3 ligase activity toward Mdm2. Ubiquitination of Mdm2 by PCAF is necessary for Mdm2 degradation and p53 activation in U2OS cells after DNA damage. 64 PCAF mediated ubiquitination appears to be regulated at least in part by the protein kinase, PKCδ. Overexpression of PKCδ is capable of inhibiting PCAF-mediated Mdm2 ubiquitination. Although the mechanism has not been fully elucidated, depletion of PKCδ results in down-regulation of Mdm2 protein expression independent of p53. 65 Taken together, these results suggest that PKCδ may regulate PCAF-mediated ubiquitination of Mdm2 during times of cellular stress.
As mentioned above, polyubiquitination of p53 by Mdm2 is necessary for proteosomal degradation. However, Mdm2 catalyzes p53 mono- or multiple monoubiquitinations in vitro and in vivo, which is not sufficient for proteasomal degradation.66-69 This suggests that additional ubiquitin ligases or co-factors are required for Mdm2-mediated p53 polyubiquitination and degradation. A yeast 2-hybrid screen recently showed that Ube4b, which functions as an E3 and E4 ubiquitin ligase, physically interacts with p53 and Mdm2 to facilitate Mdm2 polyubiquitination and subsequent degradation of p53. 70 Transient overexpression of Ube4b demonstrates that it negatively regulates the stability and function of p53 through its E4 activity in vivo and in vitro. Interestingly, UBE4B is overexpressed in brain tumors, 70 suggesting this may be a mechanism by which Mdm2 overexpression contributes to glioblastoma formation. Since UBE4B facilitates Mdm2-mediated polyubiquitination of p53, inhibiting the Mdm2:UBE3B interaction should stabilize p53 in vivo. Further investigation is warranted to determine whether inhibiting this interaction will prove useful for cancer therapies.
Since tight regulation of the antiproliferative and proapoptotic function of p53 is essential for cell viability, cells use several mechanisms to ensure proper Mdm2 expression and p53 activity during embryonic development and homeostasis. One mechanism involves Mdm4, a homolog of Mdm2 that negatively regulates p53 by binding. 71 Accordingly, genetic ablation of Mdm4 in mice leads to early embryonic lethality that is rescued by concomitant deletion of p53. 72 Mdm2 and Mdm4 interact with each other through their RING domains, located at C-termini (Fig. 2). 73 Genetic disruption of this interaction in mice revealed that Mdm2-Mdm4 heterodimerization is essential for inhibiting lethal p53 activity during embryogenesis. However, this interaction is dispensable for regulating p53 activity and Mdm2 stability in the adult mouse.74,75 These results indicate that although the Mdm4:Mdm2 interaction is important for maintaining low levels of p53 during embryonic development, Mdm2 regulation of p53 is sufficient in an adult mouse.
Mdm2 also binds the LIM domain protein, Enigma, to regulate p53 stability and activity in unstressed cells. Endogenous Enigma co-immunoprecipitates with endogenous Mdm2 in the presence or absence of p53, forming a ternary complex. The C terminus of Mdm2 was sufficient for its binding to Enigma (Fig. 2). This interaction functions to inhibit Mdm2 self-ubiquitination and enhances Mdm2-mediated ubiquitination of p53. Interestingly, Enigma was co-expressed with Mdm2 in 10 cases of human liver and stomach tumors where p53 was undetectable and in 7 cases where p53 was detected. Tissue arrays revealed that Enigma co-localized with Mdm2 in the cytoplasm of human stomach and colorectal tumor cells. 76 The Enigma-Mdm2-p53 ternary complex was detected in both the cytosolic and nuclear fractions, suggesting that Enigma regulation of Mdm2 may function to decrease p53 activity mostly in the cytosol in proliferating cells but may also suppress p53 transactivation in the nucleus. 76 This is interesting given the finding that p53 activates the proapoptotic protein, BAX, in the cytoplasm.77-79 Taken together, these results indicate that Enigma may inappropriately stabilize Mdm2 in some human tumors, resulting in destruction of p53.
Gankyrin is another protein that regulates Mdm2 mediated ubiquitination and degradation of p53. Gankyrin, which is an ankyrin repeat oncoprotein overexpressed in hepatocellular carcinomas, interacts with the S6 proteasomal ATPase to increase degradation of the tumor suppressor Rb. Similarly, U2OS cells stably overexpressing Gankyrin exhibit decreased stability of endogenous p53 protein only in the presence of Mdm2, indicating that gankyrin enhanced Mdm2 mediated degradation of p53. Co-immunoprecipitation experiments demonstrate that endogenous gankyrin physically interacts with Mdm2 but not with p53, and ubiquitination assays show that gankyrin increases Mdm2 mediated mono- and polyubiquitination of p53. 80
A genetic screen revealed that following oncogenic and oxidative stress, Seladin-1, which is activated by Ras pathway signaling, directly binds p53 and Mdm2 to regulate p53 expression. 81 Co-immunoprecipitation assays reveal binding between Seladin-1 and the N-terminus of p53, which displaces Mdm2 from p53, resulting in p53 accumulation. These results indicate that Seladin-1 regulates p53 stability by binding to Mdm2 and interfering with Mdm2-mediated p53 ubiquitination. In addition, Seladin-1 binds to Mdm2 in p53-null MEFs, 81 indicating that the Seladin-Mdm2 interaction may have p53-independent functions. The functional relevance of this interaction on additional Mdm2 targets warrants further investigation.
The majority of Mdm2 binding partners mentioned thus far regulate the MDM2-p53 feedback loop during homeostasis and in response to DNA damage. This feedback loop is also regulated in response to ribosomal stress or nucleolar stress. 82 Ribosome biogenesis involves expression of ribosomal RNA and ribosomal proteins (RPs), processing of rRNA, and subsequent assembly of the ribosome subunits in the nucleolus. The subunits then translocate to the cytoplasm, where they undergo further assembly and ultimately catalyze protein synthesis. 83 Ribosome biogenesis is the most energy demanding cellular process, and disruption of any step results in “nucleolar stress.” This triggers binding of several RPs (RPS3, 84 RPS7,85,86 RPS14, 87 RPS27, 88 RPL5, 89 RPL11, 90 RPL23, 91 RPL26 92 ) to MDM2 (Fig. 2), inhibiting its binding to and degradation of p53, resulting in p53 stabilization and activation (Table 1). Given that these Mdm2-interacting RPs all regulate the p53 response to nucleolar stress in a similar manner, it is possible that each RP may regulate different steps of the nucleolar stress response. In addition, it is possible that a combinatorial effect of RP binding to Mdm2 is required for a strong stress response. These ideas can be tested in vivo using mouse models or small molecule inhibitors that prevent binding of certain RPs to Mdm2.
As mentioned above, Mdm2 regulates the p53 family member, p73. Mdm2 binds to p73 and suppresses transcription without targeting it for degradation.93,94 Jeong et al. 95 described a novel interaction between Mdm2 and the p73 binding protein, p19ras. DNA binding assays show that P19ras interacts with both the DNA Binding Domain (DBD) and oligomerization domain of p73, but the strongest binding occurs at the DBD. P19ras and p73 co-localized in the nucleus, and this interaction resulted in increased transcriptional activation of p73. Co-precipitation experiments revealed that p19ras directly interacts with Mdm2 in a p73- and p53-independent manner. Interestingly, reporter assays demonstrate that p19ras abolishes the Mdm2-mediated transcriptional inhibition of p73. These results suggest that p19ras can alleviate the Mdm2-mediated transcriptional repression of p73 through direct binding to Mdm2. 95
Downstream Targets of Mdm2
Ubiquitin-Dependent Proteasomal Degradation
Since Mdm2 is an E3 ubiquitin ligase, the majority of Mdm2 targets, including p53, are ubiquitinated by Mdm2 and targeted for proteasomal degradation. A complete list of these proteins is provided in Table 2. Below we discuss in more detail the nature and significance of the reported interactions.
Immunoprecipitation experiments in U2OS cells reveal that in the absence of cellular stress, Mdm2 binds to and ubiquitinates the heterogeneous nuclear ribonucleoprotein K (hnRNP K), resulting in its proteasomal degradation. In response to cellular stress, however, Mdm2 is inhibited in an ATM dependent manner, leading to the rapid accumulation of hnRNP K. Interestingly, hnRNP K also functions as an important co-factor for p53 in response to DNA damage, as evidenced by decreased activation of p53 target genes after hnRNP K depletion. 96
Under normal growth conditions, Mdm2 also interacts with and ubiquitinates the p53 inducer, dual specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2), resulting in its degradation. However, under genotoxic stress, ATM phosphorylates DYRK2, enabling it to escape Mdm2-mediated degradation, resulting in its translocation to the nucleus, where it phosphorylates p53, inducing apoptosis. 97
The activating transcription factor 3 (ATF3) is a stress sensor whose induction is important for cellular responses to DNA damage. During times of normal growth, Mdm2 binds to the ATF3 leucine-zipper domain and ubiquitinates ATF3, resulting in its degradation. Importantly, deregulation of ATF3 is documented to contribute to tumorigenesis, presumably through lack of appropriate DNA damage responses. 98
Protein phosphatase 2A (PP2A) is a serine-threonine phosphatase involved in the DNA damage response. PP2A is a heterotrimer comprised of a catalytic C subunit, a structural A subunit, and several regulatory B subunits. Recent work has demonstrated that PP2A may function as a tumor suppressor (reviewed in Wurzenberger & Gerlich 99 ). Specifically, the PP2A B56γ3 isoform inhibits cell proliferation in human lung cancer cell lines. 100 Under normal growth conditions, p53 is phosphorylated by TAF1 at Thr55, rendering it inactive, but after DNA damage, B56γ-PPA dephosphorylates p53 at this residue, leading to its activation. However, phosphorylation of p53 at Ser15 by ATM is required for B56-p53 interaction and subsequent dephosphorylation at Thr55, indicating that ATM phosphorylation at Ser15 primes p53 for its interaction with B56-PPA. 101 Since B56γ3 can activate p53 during times of normal cell growth, B56γ3 must be negatively regulated, and this is achieved through Mdm2 mediated ubiquitination and proteasomal degradation. After DNA damage, B56γ3 is phosphorylated by ATM, which blocks Mdm2-mediated B56γ3 ubiquitination. ATM also phosphorylates Mdm2 at serine 395 after DNA damage, preventing Mdm2 polyubiquitination of p53.50,102 Thus, multiple mechanisms ensure p53 activity after DNA damage.
The androgen receptor (AR) is a phosphoprotein that regulates a variety of biological functions including prostate cell growth and apoptosis via the p53 pathway. Therefore, AR levels must be tightly regulated, and defects in this regulation are implicated in prostate tumorigenesis. One mechanism for regulating AR levels involves the Mdm2 and Akt pathway. 103 Mdm2, AR, and Akt form a complex in vivo in LNCaP cells (an androgen sensitive human prostate adenocarcinoma cell line that expresses endogenous Mdm2, AR, and Akt). This interaction results in the ubiquitination and proteasomal degradation of AR. 103 As previously mentioned, Akt phosphorylates Mdm2 at Ser 166 and Ser 186. 104 Mutation of these binding sites inhibits Mdm2 mediated ubiquitination and degradation of AR, indicating that Akt phosphorylation of Mdm2 is necessary for Mdm2-mediated degradation of AR. In addition, Akt phosphorylates AR itself, and this is also necessary for recruitment of Mdm2 to AR and its subsequent degradation. 103 Taken together, these data demonstrate that Akt phosphorylates AR and Mdm2, leading to Mdm2 binding to AR, which results in AR ubiquitination and subsequent degradation. Since AR expression and activity are linked to the development of prostate cancer, 105 these results provide insight into the mechanism underlying AR degradation and have important implications for prostate cancer treatments.
The homeodomain-interacting protein kinase, HIPK, is a serine-threonine kinase that functions to repress homeodomain transcription factors. As mentioned, after DNA damage, p53 induces cell cycle arrest, senescence, or apoptosis. p53 phosphorylates HIPK2 only after severe, unrepairable DNA damage, such as after UV radiation, resulting in apoptosis. However, during times of moderate DNA damage using sublethal doses of doxorubicin, the HIPK2 apoptotic activator must be silenced, and this is achieved through Mdm2 mediated ubiquitination and degradation of HIPK2. 106
The insulin-like growth factor 1 receptor (IGF-1R) is a receptor tyrosine kinase that is implicated in several cancers as a result of its antiapoptotic properties. Mdm2 physically associates with and ubiquitinates IGF-1R in vitro and in vivo, resulting in its degradation. In contrast, cells with strong p53 expression (malignant melanoma cells expressing either wild-type or mutant p53 and UV-irradiated cells) exhibit high IGF-1R expression, which could result in survival of tumor cells. 107
Notch receptors and ligands function during development and are implicated in tumorigenesis. The Notch4 receptor, which promotes tumorigenesis in mouse mammary epithelium, 108 was recently shown to be a target of Mdm2 ubiquitination. Co-immunoprecipitation experiments in p53-null H1299 cells, which endogenously express NICD4 and Mdm2, reveal that NICD4 is a direct Mdm2 target and that this interaction is p53-independent. 109 Interestingly, Mdm2 also ubiquitinates Numb, an antagonist against Notch signaling, leading to its proteasomal degradation. 110
As discussed in the previous section on Mdm2 effectors, several ribosomal proteins (RPs) bind to Mdm2, inhibiting its binding to p53. This ultimately results in p53 stabilization and activation. However, the interaction between Mdm2 and RPL26 performs a different function. Mdm2 acts as a ubiquitin ligase for RPL26, resulting in its degradation. 111 Since RPL26 binds to the 5′UTR of p53 to increase its translation, 112 in this scenario, Mdm2 inhibits p53 translation via RPL26 degradation. These data indicate that under nonstressed conditions, Mdm2 binds to RPL26 to keep p53 levels low. However, in response to stress, Mdm2 ubiquitination of RPL26 is inhibited by unknown mechanisms, resulting in increased p53 expression. These results reveal an additional mechanism used by cells to regulate p53 expression in stressed versus normal cells.
RPS7 is another substrate for Mdm2 ubiquitination. 86 Since RPS7 can also bind Mdm2, inhibiting its interaction with p53, RPS7 is both a substrate and effector of Mdm2. 86 Finally, RPS27L, an RPS27-like protein, is also a substrate of Mdm2 ubiquitination. 88 Although both family members (RPS27L and RPS27) bind to Mdm2, only RPS27L can be degraded by Mdm2 under physiological conditions. Since RPS27L competes with p53 for binding to Mdm2, overexpression of this protein results in accumulation of p53. Given that RPS27L is a direct p53 target,113,114 these data demonstrate a feedback loop wherein in response to stress, p53 activates RPS27L, which then competes with p53 for Mdm2 binding, resulting in p53 stabilization. However, p53 also induces Mdm2, which degrades both p53 and RPS27L. Together, these results reveal a complex relationship between RPS27L, Mdm2, and p53 that could have important implications for regulation of cell proliferation under stressed conditions.
Ubiquitin Ligase Independent Functions of Mdm2
Mdm2 has E3-ligase independent functions as observed with Mdm2 dependent transcriptional activation of the NFκB subunit p65. p65 is an antiapoptotic factor expressed in neoplastic cells, namely leukemic bone marrow cells. Chromatin immunoprecipitation and EMSA demonstrate that Mdm2 directly binds the Sp1-binding site of the p65 promoter. Interestingly, Mdm2 overexpression in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) and an ALL cell line (EU-4) results in increased expression of p65 and resistance to doxorubicin. Together, these results implicate Mdm2 activation of p65 in chemotherapy resistance in ALL. 115
Mdm2 can also induce translation of the Inhibitor of Apoptosis (IAP) protein family member, XIAP. XIAP is a caspase inhibitor that is overexpressed in cancer cells and confers resistance to DNA damage induced by irradiation and chemotherapy (reviewed in Galban & Duckett 116 and Dubrez-Daloz et al. 117 ). Overexpression of Mdm2 is also implicated in resistance to DNA damage in a p53-independent manner. 115 The mechanism behind this resistance was recently elucidated when Mdm2 was demonstrated to positively regulate IRES-dependent XIAP translation during cellular stress. 118 Previous work demonstrated a link between Mdm2 and XIAP when overexpression of Mdm2 in a p53-null leukemia cell line resulted in XIAP protein upregulation. 115 EU-1 cells (acute lymphoblastic leukemia cell line that overexpresses Mdm2) were treated with IR, which resulted in decreased nuclear Mdm2 expression and increased Mdm2 cytoplasmic expression. Metabolic [ 36 S]-methionine labeling and IP analysis of these cells revealed a significant increase in metabolically labeled, newly synthesized XIAP RNA in the IR-treated cells. In contrast, protein levels were unchanged in IR treated cells compared with untreated cells, indicating that induction of XIAP by IR occurs at the translational level. To determine whether Mdm2 directly mediates XIAP translation, Gu et al. 118 performed an RNA binding assay using glutathione S-transferase (GST) fusion proteins containing either full length Mdm2 or deletion mutants. These experiments revealed that the C-terminal RING domain (amino acids 425-491) of Mdm2 physically interacts with the XIAP IRES both in vitro and in vivo. Since XIAP is upregulated in IR-resistant cancer cells, Gu et al. 118 examined the mechanism underlying this resistance and found that overexpression of Mdm2 increases resistance to IR-induced apoptosis. Conversely, blocking the interaction between Mdm2 and XIAP confers sensitivity. 118 These data demonstrate that Mdm2 positively regulates XIAP translation in response to IR. Taken together, these results have important implications for tumors that are resistant to IR-induced apoptosis and reveal a novel role for Mdm2 in tumorigenesis that is independent of p53.
Nbs1 is a member of the Mre11/Rad50/Nbs1DNA repair complex that functions in double strand break repair, meiotic recombination, and telomere maintenance. 119 Mutations in Nbs1 result in genetic instability syndromes such as Nijmegen breakage syndrome (NBS) and have a very high incidence of cancer as a result of defective DNA repair.120-122 Mdm2 directly binds to Nbs1 and Mdm2-Nbs1 co-localized to DNA damage sites following gamma-radiation in HeLA and IMR90 cells, inhibiting DNA double strand break repair as measured by a comet assay. Moreover, the ubiquitin ligase domain of Mdm2 was dispensable for its binding to Nbs1, and this interaction was p53 independent as it was observed in p53-null MEFs. 123
p53 Independent Functions of Mdm2
As demonstrated by the p53-independent function of Mdm2 in inhibiting DNA double strand break repair, Mdm2 has p53-independent functions. For example, in a mouse model of breast cancer, high Mdm2 protein levels resulted in chromosomal abnormalities in a p53-independent manner. 124 Interestingly, the tumor spectrum of Mdm2 transgenic mice differs from that observed in p53-null mice, suggesting that Mdm2 has p53-independent functions during tumorigenesis. 14 The elevated Mdm2 levels in these transgenic mice induce chromosome breaks and aneuploidy in developing and mature B cells in a p53-independent manner. 125 Lastly, overexpression of an MDM2 cDNA induces G0/G1 arrest in a p53-independent manner in normal human and mouse cell lines. 126 Since Mdm2 is overexpressed in human tumors, 10 these data suggest that tumor cells bypass this cell cycle arrest by unknown mechanisms and go on to become tumors.
Concluding Remarks
Although the most documented function of Mdm2 involves its negative regulation of p53, the discovery of new Mdm2 binding partners and substrates demonstrates that Mdm2 has many functions in addition to regulating p53. Analysis of the Mdm2 binding partners presented in Tables 1 and 2 demonstrates that transcriptional, post-transcriptional, and post-translational regulation of Mdm2 is important to facilitate appropriate Mdm2 levels during development, in homeostasis, and in response to DNA damage. Importantly, errors in the transcriptional regulation of Mdm2 or post-translational modifications of Mdm2 can contribute to tumorigenesis in a p53-independent manner. Mouse models harboring genetic alterations in Mdm2 binding proteins and/or amino acid substitutions in Mdm2 phosphorylation sites will reveal the physiological relevance of these reported interactions. Understanding how Mdm2 activities are regulated in vivo will be enormously valuable for developing treatments for cancers that retain wild-type p53.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
NIH/NCI (T32 CA009299-32 and R01 CA47296-24) to G.L.