
Abstract
The MDM2 oncogene is a key negative regulator of the p53 tumor suppressor protein. MDM2 and p53 form an autoregulatory feedback loop to tightly control the proper cellular responses to various stress signals in order to prevent mutations and tumor formation. The levels and function of the MDM2 protein, an E3 ubiquitin ligase, are regulated by a wide variety of extracellular and intracellular stress signals through distinct signaling pathways and mechanisms. These signals regulate the E3 ubiquitin ligase activity of MDM2, the ability of MDM2 to interact with p53 and a number of other proteins, and the cellular localization of MDM2, which in turn impact significantly upon p53 function. This review provides an overview of the regulation of MDM2 activities by the signals and factors that regulate the MDM2 protein, including genotoxic stress signals, oncogenic activation, cell cycle transition, ribosomal stress, chronic stress, neurohormones, and microRNAs. Disruption of the proper regulation of the MDM2-p53 negative feedback loop impacts significantly upon the frequency of tumorigenesis in a host. A better understanding of the complex regulation of MDM2 and its impact upon p53 function in cells under different conditions will help to develop novel and more effective strategies for cancer therapy and prevention.
Keywords
Introduction
MDM2 is an oncogene that was discovered in a locus amplified on double minute chromosomes in a tumorigenic mouse cell line (3T3-DM). 1 The main function of MDM2 is to negatively regulate the levels and function of p53 tumor suppressor proteins. High MDM2 levels decrease p53 protein levels and attenuate p53 function, which increase cancer risk and/or accelerate tumor formation and progression. Overexpression of MDM2 is observed in some human tumors. Amplification of the mdm2 gene is a major mechanism of MDM2 overexpression. Furthermore, MDM2 gene amplification and the mutation of p53 are most commonly mutually exclusive. 2 In addition, SNP309, a naturally occurring polymorphism in the mdm2 gene, leads to the increased MDM2 transcriptional levels in humans, which is associated with increased risk for several cancers. 3
MDM2 is a member of the RING finger family of E3 ligases that contains several conserved functional regions: the N-terminal region contains a p53 binding domain; the central region contains nuclear localization and export sequences, an acidic domain, a zinc finger domain, and binding sites for TBP, 4 p300,5,6 and ARF 7 ; and the C-terminal region contains a RING finger domain (Fig. 1). The RING finger domain binds to an E2 ubiquitin-conjugating enzyme to promote the ubiquitination of target proteins. The main substrate of MDM2 is p53, although MDM2 also ubiquitinates other substrates such as MDM4, β-arrestin, NUMB, ribosomal protein S7, PCAF, and the insulin-like growth factor 1 receptor.8-12 MDM2 binds to p53 and mono-ubiquitinates and poly-ubiquitinates p53. Poly-ubiquitination of p53 marks p53 for proteasomal degradation. The interaction of MDM2 with p53 can mediate the translocation of p53 to the cytoplasm, 13 thereby removing it from its nuclear site of action and leading to rapid p53 degradation by cytoplasmic proteasomes. In addition, MDM2 negatively regulates p53 function by binding to the p53 transactivation domain to prevent its activity. This binding of MDM2 to the p53 N-terminal transactivation domain prevents the interaction of p53 with the basal transcription machinery.14-16 The critical role of MDM2 in the negative regulation of p53 is best illustrated by elegant mouse studies; mice deficient for MDM2 have an embryonic lethal phenotype due to excessive p53-dependent apoptosis, which can be rescued by knocking out the p53 gene. 17

Schematic model showing the domain structure of the MDM2 protein. Functional domains of the MDM2 protein include the p53 binding domain, nuclear localization sequence (NLS), nuclear export sequence (NES), acidic domain, zinc finger domain, and RING finger domain. The amino acid residues are numbered.
The negative regulation of p53 by MDM2 can be modulated by another member of the MDM2 family, MDM4. The amino acid sequences of MDM4 are highly homologous to MDM2; MDM4 also contains an N-terminal p53 binding domain, a central acidic domain, and a C-terminal RING finger domain. MDM4 can bind to p53 and block the p53 transcriptional activity. While MDM4 does not stimulate p53 degradation through direct ubiquitination, MDM4 forms heterodimers with MDM2 through its RING finger domain, which then influences the E3 ubiquitin ligase function of MDM2.18-20 Depending upon the circumstances, MDM4 either enhances or inhibits the E3 ubiquitin ligase function of MDM2 for the p53 protein.20-22 MDM4 can be overexpressed in human tumors, and the spectrum of tumors with MDM4 overexpression is different from that with MDM2 overexpression. 23 Loss of MDM4 also leads to p53-dependent embryonic lethality in mice, but the fetus dies at different times during development when MDM2 or MDM4 is deleted. 24 Therefore, MDM4 acts as a critical regulator of the p53-MDM2 feedback loop.
As a key negative regulator of p53 protein levels and activity, MDM2 is a highly regulated protein. MDM2 is a transcriptional target of p53. p53 positively regulates MDM2 through binding to the p53 DNA consensus binding element in the first intron of the mdm2 gene to form an autoregulatory negative feedback loop with MDM2.25-27 Stress-mediated p53-dependent increased levels and activity of MDM2 may play an important role in the regulation of the duration and amplitude of the p53 response after stress. In addition, various extracellular or intracellular signals function through distinct signaling pathways to regulate MDM2 levels, activity, and intracellular localization, leading to the activation or inhibition of p53 function. Here, we review the regulation of MDM2 and its impact upon p53 function by these signals and factors, including DNA damage, oncogenic activation, cell cycle transition, ribosomal biogenesis, chronic stress, neurohormones, and microRNAs.
The Regulation of MDM2 by Genotoxic Stress Signals
DNA Damage (Ionizing Radiation and Ultraviolet Light)
In response to ionizing radiation (IR), p53 is rapidly activated by the ATM protein kinase, leading to the accumulation of p53 proteins in cells. The phosphorylation of p53 at Ser-15 by ATM, which reduces the affinity of p53 for MDM2, results in reduced p53 degradation by MDM2 and thus enhanced p53 protein stability and function. The rapid ATM-dependent MDM2 phosphorylation on Ser-395 prior to p53 protein accumulation also plays a critical role in this process. The phosphorylation of MDM2 Ser-395 blocks the ability of MDM2 to export p53 from the nucleus, 28 which prevents p53 degradation and promotes the accumulation of p53 proteins in cells. In addition, the phosphorylation of MDM2 at Ser-395 also reduces the RING domain oligomerization, which in turn attenuates the processivity of the E3 ligase activity of MDM2. 29 The phosphorylation of MDM2 at Ser-395 also promotes an interaction between p53 mRNA and the MDM2 protein, which increases the synthesis of the p53 protein after genotoxic stress. This p53 mRNA–MDM2 interaction also promotes SUMO-conjugation of MDM2 and its accumulation in the nucleoli compartment, preventing the negative regulation of p53 by MDM2 and in turn leading to the activation of p53 in the nucleoplasm.30-32 ATM not only directly phosphorylates MDM2 but also activates a second kinase, c-Abl, to indirectly regulate MDM2 in response to genotoxic stress. 33 c-Abl interacts with MDM2 and phosphorylates MDM2 at multiple sites, reducing the ability of MDM2 to down-regulate p53 (Fig. 2). In addition, ATM regulates HAUSP, a specific de-ubiquitinase for both p53 and MDM2, in response to genotoxic stress. In nonstressed cells, HAUSP is phosphorylated at Ser-18, which maintains the stability of HAUSP and prevents auto-ubiquitination of MDM2 through interaction with MDM2. 34 In response to DNA damage, ATM activates PPM1G, a phosphatase that dephosphorylates HAUSP at Ser-18 and enhances HAUSP degradation, which in turn increases the degradation of MDM2 and the accumulation of p53 proteins in cells. 35
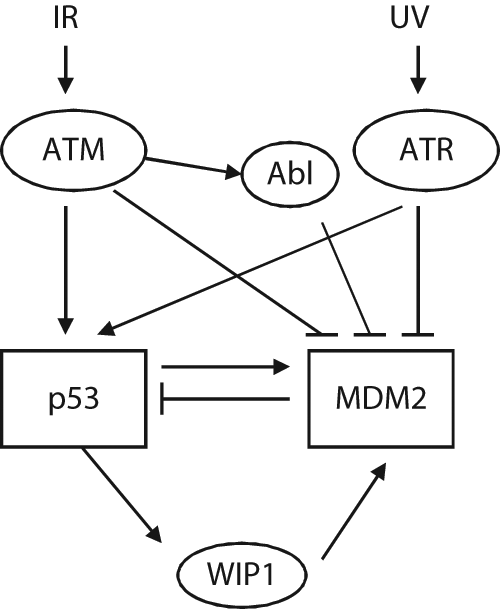
The regulation of MDM2 by DNA damage. Ionizing radiation (IR) and ultraviolet (UV) light activate ATM and ATR, respectively. Activated ATM and ATR phosphorylate p53 and MDM2, which result in the decreased function of MDM2 and stabilization of the p53 protein. In addition, ATM activates Abl to phosphorylate and down-regulate MDM2 function. p53 activation leads to transcriptional induction of its target genes. Wip1, a p53 target gene, is a phosphatase that dephosphorylates MDM2 at the site phosphorylated by ATM, leading to the increase of MDM2 function and activity.
Ultraviolet (UV) light exposure is another type of radiation that rapidly activates p53, leading to the accumulation of the p53 protein. UV light exposure preferentially activates the ATR protein kinase, which phosphorylates both p53 and MDM2. ATR phosphorylates p53 at multiple sites, including Ser-15 and Ser -37, and phosphorylates MDM2 at Ser-407.36,37 The phosphorylation of MDM2 at Ser-407 by ATR reduces MDM2-dependent export of p53 from nuclei to cytoplasm, which in turn increases the accumulation of the p53 protein and its function 37 (Fig. 2).
Thus, at the early stage of cellular response to genotoxic stress, the modifications of both p53 and MDM2 through different mechanisms are responsible for the rapid stabilization of the p53 protein and activation of the p53 signaling pathway. p53 then exerts its function to maintain genomic stability through transcriptional regulation of its target genes. Meanwhile, the activation of p53 also leads to increased MDM2 expression. This delayed increase of MDM2 reduces the p53 protein levels. This results in oscillations of MDM2 and p53 levels that are temporally out of phase with each other. This could maintain a proper p53 response toward stress in cells, reducing the risk of having too much p53 and leading to cell death when cell cycle arrest and DNA repair are a more appropriate response.38,39 In addition, Wip1, a p53 target gene and a phosphatase that is induced by p53 at a later time in response to genotoxic stress, can dephosphorylate MDM2. 40 Wip1 dephosphorylates MDM2 at Ser-395, the site phosphorylated by ATM. Dephosphorylation of MDM2 at Ser-395 increases the affinity of MDM2 toward p53 to enhance p53 ubiquitination and degradation and at the same time decreases MDM2 auto-ubiquitination (Fig. 2). Thus, Wip1 acts as an additional important regulator for the p53-MDM2 negative feedback loop.
The Regulation of MDM2 by Oncogenes
Oncogenic Activation
Aberrant activation of a number of oncogenes (E2F-1, β-catenin, Myc, Ras, nucleophosmin, etc.) can activate p53, which is crucial for tumor suppression. One mechanism of this p53 activation involves the p14/p19 ARF tumor suppressor protein that inhibits MDM2. ARF is the alternate reading frame protein that is encoded by the Ink4a locus. 41 The transcription of ARF is induced by E2F-142 and β-catenin, 43 and its transcription is repressed by p53. In addition, Myc, Ras, and nucleophosmin increase the ARF protein levels by inhibiting the degradation of the ARF protein.44-46 ARF is ubiquitinated and degraded through binding to ULF (ubiquitin ligase for ARF). Myc inhibits ULF-mediated ARF ubiquitination through interaction with ULF. Nucleophosmin overexpression leads to co-localization of ARF and nucleophosmin in nucleoli, segregating ARF from its ubiquitin ligase that is mainly present in the nucleoplasm. 47 ARF accumulates in nucleoli of cells. ARF binds to the central domain of the MDM2 protein and promotes the accumulation of MDM2 in the nucleoli, resulting in the segregation of MDM2 from p53. The interaction of ARF with MDM2 also antagonizes MDM2’s E3 ubiquitin ligase activity toward p53, which prevents MDM2-mediated p53 degradation and increases p53 levels and activity31,48 (Fig. 3). Disruption of this ARF-MDM2-p53 tumor surveillance pathway predisposes an individual to cancer, and inactivation of INK4a-ARF by deletion, silencing, or mutation has been frequently observed in many human cancers. 49
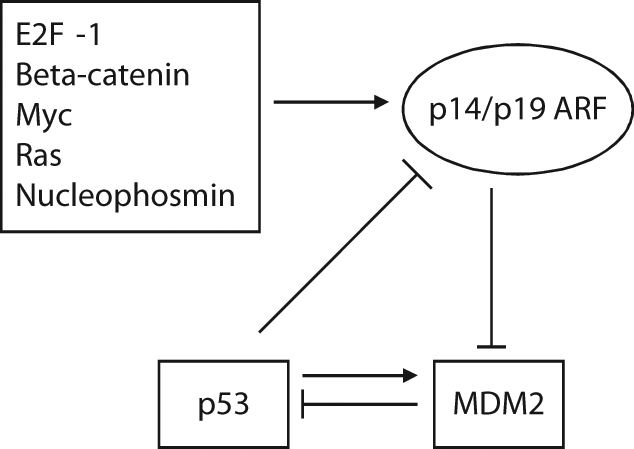
The ARF-MDM2-p53 loop. Aberrant activation of a number of oncogenes increases ARF transcription or protein levels. ARF binds to MDM2 to accumulate MDM2 in nucleoli and inhibit its E3 ubiquitin ligase activity toward p53, which leads to p53 protein accumulation and increased p53 function. The transcription of ARF can be down-regulated by p53 itself.
Other Oncogenes
AKT and survival signaling
IGF-1/AKT pathway is an evolutionally conserved pathway that plays critical roles in the regulation of cell proliferation and survival. The binding of IGF-1 to its tyrosine kinase receptor (IGF-1R) results in the activation of the PI3 kinase (PI3K), which in turn phosphorylates the phosphoinositides and leads to increased PIP3 levels at the plasma membrane. PIP3 then recruits protein kinases containing pleckstrin homology domains to the membrane, including AKT, PDK1, and PDK2, 2 upstream activators of AKT. Increased PIP3 activates PDKs, which then phosphorylate AKT at Thr-308 and Ser-473.50-52 The activation of AKT permits the release of AKT from the membrane to interact with and phosphorylate a range of cytoplasmic and nuclear substrates, which leads to the inhibition of apoptosis and promotion of cell survival. MDM2 is a substrate of AKT. AKT phosphorylates MDM2 at Ser-166/186, both of which lie within the RXRXXS/T consensus motifs for AKT kinase. 53 The phosphorylation of MDM2 at Ser-166/186 promotes its nuclear localization and its interaction with p300, a transcriptional co-activator that forms a complex with MDM2 and promotes p53 degradation.5,54 This also inhibits the interaction of MDM2 with ARF. Therefore, the phosphorylation of MDM2 at Ser-166/186 by AKT increases the activity of MDM2, which results in increased p53 degradation and the inhibition of p53 function (Fig. 4).
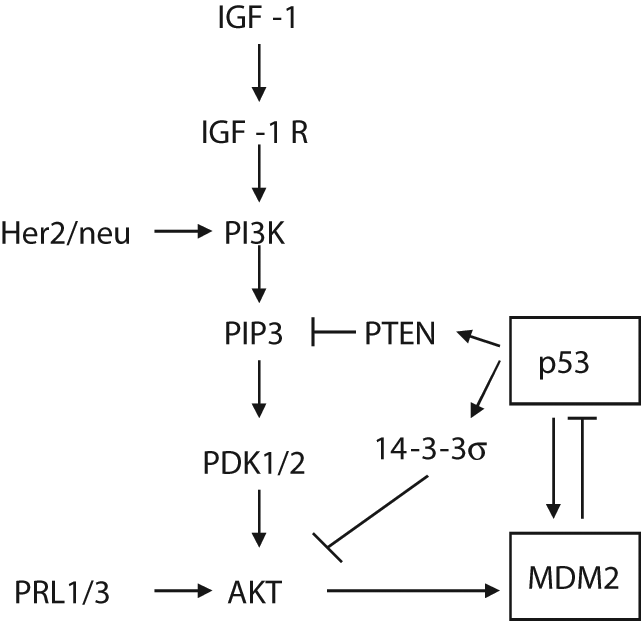
The activation of MDM2 by the AKT pathway. AKT phosphorylates and activates MDM2, which promotes the degradation of the p53 protein. The AKT activity and its regulation of MDM2 are positively or negatively regulated by a number of factors. PTEN and 14-3-3σ, 2 transcriptional targets of p53, inhibit AKT activity and its regulation of MDM2, which in turn enhances p53 function. PRL-1, PRL-3, and Her2/neu activate the AKT pathway, which in turn inhibits p53 function.
The AKT activity and its regulation of MDM2 and p53 are positively or negatively regulated by a number of factors (Fig. 4). Two p53 transcriptional targets, PTEN and 14-3-3σ, have been shown to inhibit the AKT activity and increase p53 function through down-regulation of MDM2.55,56 PTEN is a PIP3 phosphatase that degrades PIP3 to PIP2, which no longer activates PDKs, therefore decreasing the AKT activity. 14-3-3σ directly binds to AKT and inhibits its activity. In turn, both PTEN and 14-3-3σ reduce the phosphorylation of MDM2 at Ser-166/186 and increase the p53 activity by preventing MDM2-dependent p53 degradation. Therefore, both PTEN and 14-3-3σ connect the p53 pathway with the AKT pathway and form a positive feedback loop to enhance the p53 activity and down-regulate the AKT activity. In addition, tyrosine phosphatases from regenerating liver 1 and 3 (PRL-1 and PRL-3) have been shown to enhance AKT activity, resulting in the phosphorylation of MDM2. 57 Her-2/neu, which is often amplified or overexpressed in human cancers, can activate the AKT pathway, which in turn enhances MDM2 phosphorylation and inhibits p53 function. 53 The AKT activity has been found to be elevated in many human tumors. 58 The aberrant activation of the AKT kinase can result from the amplification of the AKT gene. In cancers, it is also common to observe the altered regulation of the downstream pathways under AKT regulation caused by the amplification of Her-2/neu, mutations of the catalytic subunit of PI3K, loss of the PTEN gene or protein, and loss of 14-3-3σ expression.56,59,60
The Regulation of MDM2 during Cell Cycle Transition
It has been shown that cell cycle transition may regulate the activity of MDM2. 61 The murine MDM2 protein contains a cyclin recognition motif that is located between the nuclear localization and nuclear export sequences with a sequence of RRSL (residues 181-184). Cyclin A–CDK1 and cyclin A–CDK2, but not other cyclin-containing complexes, bind to MDM2 and phosphorylate MDM2 at Thr-216. The phosphorylation of MDM2 at Thr-216 modestly reduces the p53-MDM2 interaction. It is possible that because Thr-216 is outside of the p53-MDM2 interaction domain, the phosphorylation of MDM2 at Thr-216 has an indirect effect on the p53-MDM2 interaction by altering the conformation of the MDM2 protein. Meanwhile, the phosphorylation of MDM2 at Thr-216 modestly increases the binding of MDM2 to ARF. Both the reduced p53-MDM2 interaction and the increased MDM2-ARF interaction result in the increased activity of p53 (Fig. 5). The phosphorylation of MDM2 at Thr-216 occurs at the onset of S phase when levels of the cyclin A protein become detectable and Thr-216 phosphorylation disappears when cells pass through S phase. It has been shown that cyclin G, a transcriptional target of p53, interacts with PP2A phosphatase and stimulates the ability of PP2A to dephosphorylate MDM2 at Thr-216; thus, cyclin G–PP2A phosphatase enhances MDM2 activity and inhibits p53. 62 Cyclin G–null MEF cells have high phosphorylation levels of MDM2 Thr-216 and high p53 protein levels. 62 A possible explanation for Thr-216 phosphorylation and the resultant modest increase of p53 at the onset of S phase is that p53 has an increased transcriptional activity toward p21. p21 has been reported to bind to cyclin A– and cyclin B–containing complexes when cells pass S phase to control the activity of both cyclin A–CDKs and p53 when cells go into S phase.63-65
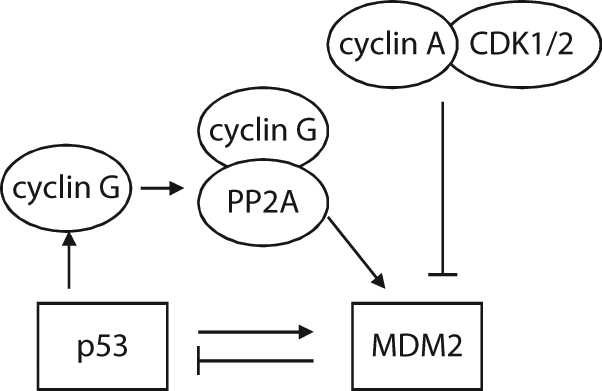
The regulation of MDM2 during cell cycle transition. Phosphorylation of MDM2 by cyclin A–CDK1/2 down-regulates the MDM2 activity, which occurs at the onset of S phase and disappears when cells pass through S phase. Cyclin G, a transcriptional target of p53, forms a complex with PP2A, which removes the phosphorylation of MDM2 by cyclin A–CDK1/2, and therefore enhances MDM2 activity.
The Regulation of MDM2 by Ribosomal Stress
The MDM2-p53 negative feedback loop is also regulated by ribosomal stress, also known as nucleolar stress. Ribosomal biogenesis is a coordinated cellular process that involves the expression of ribosomal RNA (rRNA) and ribosomal proteins (RPs), processing of rRNA, and assembly of RPs and rRNA to generate mature 80S ribosomes to ensure an adequate rate of protein synthesis to enter the cell cycle and maintain cellular homeostasis. This entire process consumes a significant amount of cellular resources and plays an important role in a number of important cellular activities. 66 Perturbations of ribosomal biogenesis, including inadequate rRNA transcription, disruption of rRNA processing, and RP imbalances, induce ribosomal stress, which activates p53 through the RP-MDM2-p53 pathway. Several RPs, including RPL5, RPL11, RPL23, RPL26, RPS3, RPS7, RPS14, and RPS27/L, have been shown to be able to interact with MDM2. MDM2 is a nucleocytoplasmic shuttling protein, and RPs mostly reside between cytosol, where they are synthesized, and the nucleoli, where they are assembled into the ribosomes. The interaction of RPs and MDM2 may occur under the following conditions: 1) the overexpression of the oncogene Myc increases the transcription of rRNAs, RPs, and tRNAs. Myc also increases the translational rate of RPs along with a global increase in the rate of translation 67 ; 2) when free RPs are released into the nucleoplasm due to the breakdown of nucleoli, which is triggered by the disruption of ribosomal biogenesis68,69; and 3) when MDM2 shuttles into nucleoli as a consequence of its interaction with the nucleolar protein ARF.31,70 The resultant excess of free RPs then binds to the central acidic region of MDM2 in a manner dictated by specific sequence requirements for binding. For example, the MDM2 C4 zinc finger region is critical for its interaction with RPL5 and RPL11 but not for RPL23. Zinc finger mutant MDM2C305F loses interaction with RPL5 and RPL11,71,72 while a slight different zinc finger mutant MDM2C305S loses interaction with RPL11 but not RPL5 and RPL23. 73 The RP-MDM2 interaction blocks the E3 ubiquitin ligase function of MDM2, which results in the accumulation and activation of p53 (Fig. 6). It is currently unclear how the binding of RPs to the central acidic region of MDM2 inhibits the E3 ubiquitin ligase function of its C-terminal RING finger domain. A possible mechanism is that the binding of RPs to the MDM2 central region reduces its flexibility, and the rigid MDM2 is thus unable to bring its RING finger domain and p53 together. In addition, RPS7 functions as both the effector and affector of MDM2. RPS7 inhibits MDM2 E3 ligase function, which can be facilitated by MDM4, and RPS7 itself is a substrate of MDM2. 12 RPS27-like protein (RPS27L), which inhibits the activity of MDM2 to activate p53, is a p53 target gene and forms a positive feedback loop with p53. 74 Interestingly, not all RP-MDM2 interactions activate p53; the interaction of RPL26 with MDM2 has a different impact upon the p53 activity. RPL26 increases the translational rate of p53 mRNA by binding to its 5′-untranslated region (UTR). 75 RPL26 is a substrate of MDM2; MDM2 binds to RPL26 to prevent the interaction of RPL26 with p53 and drives the poly-ubiquitylation and proteasomal degradation of RPL26, which leads to the inhibition of p53 translation. 76 It is currently unclear why multiple RPs bind to and interact with MDM2 to active p53 function in response to ribosomal stress. One possibility is that because different RPs bind to different amino acid sequences in the MDM2 protein, they can bind MDM2 simultaneously and have a synergistic inhibitory effect on MDM2 to fully activate p53 in cells.71,72,77 Interestingly, heterozygous mutations in several RPs that interact with MDM2, including RPL5, RPL11, and RPS7, have been found in Diamond-Blackfan anemia patients, who have chronic regenerative anemia, various degrees of congenital abnormality, and increased risk of malignancy.78,79 Mutations of many RPs in zebrafish show an association with growth impairment and tumor predisposition. 80 These findings all suggest that the RP-MDM2-p53 pathway is functional in vivo and plays an important role in monitoring proper ribosomal biogenesis.
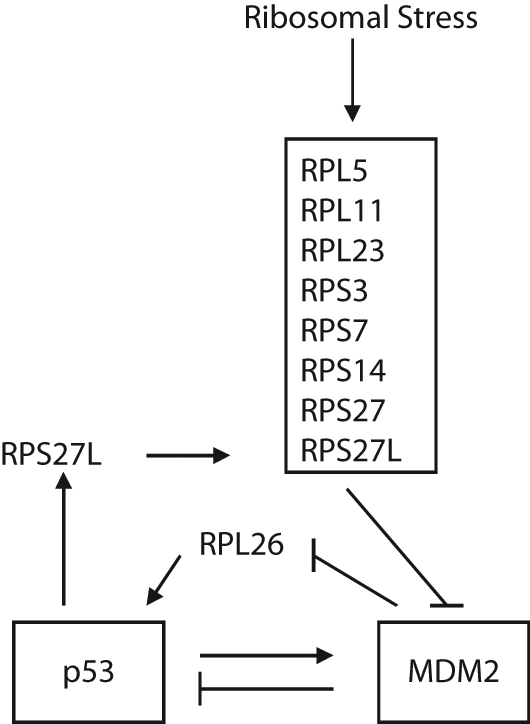
The regulation of MDM2 by ribosomal stress. Ribosomal stress can be induced by the perturbation of ribosomal biogenesis, including inadequate rRNA transcription, disruption of rRNA processing, and imbalance of ribosomal proteins (RPs). In response to ribosomal stress, a number of RPs, including RPL5, RPL11, RPL23, RPS3, RPS7, RPS14, RPS27, and RPS27L, interact with MDM2 to inhibit MDM2 function and activate p53. Among these RPs, RPS27L is a p53 target gene, which can be positively regulated by p53. In addition, RPL26 binds to 5′-UTR of the p53 gene and increases the translational rate of p53. RPL26 is also a substrate of MDM2. The interaction of MDM2-RPL26 promotes the degradation of RPL26 and inhibits p53 translation.
The Regulation of MDM2 by Chronic Stress and Neurohormones
In an intact organism, the “psychological stress response” refers to an intricate process that involves the change of information processing pathways in the central nervous system and periphery in response to environmental and psychological factors, which leads to fight or flight, or defect/withdrawal responses. 81 In response to psychological stress, 2 main systems, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic-adrenal medulla (SAM), are activated. The hypothalamus secretes corticotropin-releasing factor into a portal circulation to the anterior pituitary, which secretes adrenocorticotropic hormone (ACTH) into the general circulation, stimulating the release of glucocorticoid hormones from the adrenal cortex. The activation of the SAM increases the synthesis of norepinephrine from the locus coeruleus and epinephrine levels in the periphery. Acute activation of these pathways is necessary for adaptive processes and normally prepares humans or animals to endure a threat. However, under chronic stress, most organs are negatively affected by the prolonged activation of these pathways with extended exposure to elevated levels of these neurohormones.
Chronic stress, such as depression or lack of social support, has been shown to have significant negative influences on the onset, progression, and mortality of various cancers.82-86 A recent meta-analysis of 165 longitudinal studies demonstrated that psychosocial factors and stressful life experiences are associated with higher cancer incidence, poorer cancer survival, and higher mortality. 85 However, the molecular mechanism by which chronic stress promotes tumorigenesis is not well understood. Interestingly, recent studies suggest that neurohormones elevated during chronic stress down-regulate p53 through the activation of MDM2, which could be an important mechanism by which chronic stress promotes tumorigenesis.87,88 Glucocorticoid hormones, including cortisol (a major glucocorticoid in humans) and corticosterone (a major glucocorticoid in mice), clearly increase phosphorylation levels of MDM2 Ser-166/186 and decrease p53 protein levels and function. 87 This effect is largely mediated by serum- and glucocorticoid-induced protein kinase (SGK1), a gene regulated by glucocorticoids. Glucocorticoids bind to a glucocorticoid receptor, which then translocates to the nucleus and acts as a transcriptional factor to regulate the expression of its target genes, including SGK1.89,90 SGK1 is a ubiquitously expressed serine-threonine kinase, which shares high sequence homology with AKT (~50% through their catalytic domain) and a similar consensus phosphorylation site RXRXXS/T with AKT. 91 Interestingly, similar to AKT, SGK1 activates MDM2 through phosphorylation of MDM2 at Ser-166/186,87,92 which in turn down-regulates p53 function (Fig. 7). Blocking SGK1 function abolishes the phosphorylation of MDM2 at Ser-166/186 by glucocorticoids and the inhibitory effect of glucocorticoids on p53 function. Consistent with these results, chronic stress with continuous elevation of glucocorticoids clearly decreases p53 function. In p53+/– mice, chronic stress greatly promotes IR-induced tumorigenesis. Furthermore, chronic stress promotes the growth of human xenograft tumors in a largely p53-dependent manner. 87
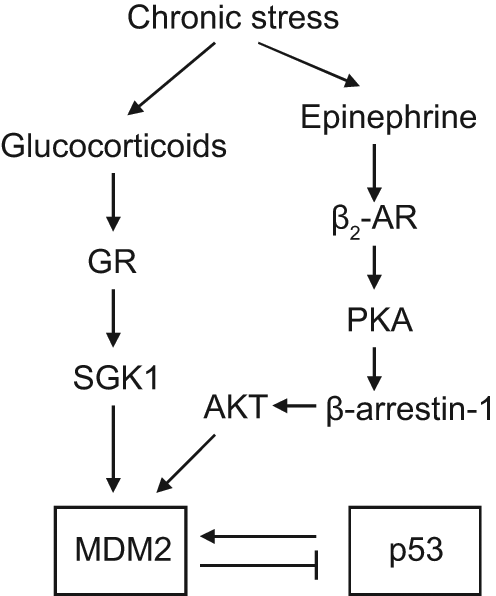
The regulation of MDM2 by chronic stress and neurohormones. Chronic stress increases the levels of neurohormones, including glucocorticoids and epinephrine, both of which activate MDM2 and inhibit p53 function through distinct signaling pathways. Glucocorticoids bind to a glucocorticoid receptor (GR) to induce the expression of SGK1, which phosphorylates and activates MDM2. Epinephrine functions through β2-adrenergic receptor (AR) to activate the PKA–β-arrestin-1 pathway, which then promotes AKT-mediated MDM2 activation.
Epinephrine and norepinephrine exert most of their functions by binding to a variety of adrenergic receptors (ARs), including α1, α2, β1, β2, and β3 receptors. 93 Prolonged treatment of isoproterenol, a synthetic analog of epinephrine, was reported to down-regulate p53 protein levels through the activation of MDM2. This effect of isoproterenol on MDM2 and p53 is through its binding to β2-ARs, which promotes the activation of PKA, followed by the recruitment of β-arrestins to activate the PKA–β-arrestin pathway. 88 In both mice and human cells, the activation of β-arrestin-1 facilitates AKT-mediated activation of MDM2. β-arrestin-1 also acts as a molecular scaffold to promote the MDM2-p53 interaction and the degradation of the p53 protein (Fig. 7). Furthermore, this prolonged treatment of isoproterenol increases DNA damage in cells, which may also promote tumorigenesis.
The Regulation of MDM2 Translation by MicroRNAs
MicroRNAs are endogenously expressed, small noncoding RNAs, which play a key role in the posttranscriptional regulation of gene products. MicroRNAs pair with partially complementary sites in 3′-UTRs of target mRNAs, leading to translational repression of target genes. Aberrant microRNA expression has been observed in human cancers.94-96 Emerging evidence demonstrates that microRNAs play an important role in tumorigenesis.97-100 Several microRNAs targeting MDM2 have been identified, including miR143/145, miR605, miR25, and miR32.101-103 miR143/145, which can be posttranscriptionally induced by p53, negatively regulates both MDM2 mRNA and protein levels through direct binding to 3′-UTR of MDM2 mRNA, resulting in the increase of p53 protein levels and function. 101 The miR605 gene is a transcriptional target of p53. Overexpression of miR605 directly decreases MDM2 protein levels and increases p53 function. 103 The transcription of miR25 and miR32 is negatively regulated by p53 and positively regulated by E2F-1 and Myc. miR25 and miR32 also negatively regulate MDM2 protein levels through direct binding to 3′-UTR of MDM2 mRNA, and this stabilizes the p53 protein and increases p53 function. 102 Overexpression of miR25 and miR32 in glioblastoma multiforme cells inhibits growth of tumor cells in the mouse brain. These microRNAs thus form feedback loops with MDM2-p53 to decrease MDM2 levels and promote p53 functions (Fig. 8). Future identification of microRNAs targeting components regulating MDM2, such as ARF, AKT, and others, will help to further understand the regulation of MDM2 by microRNAs.
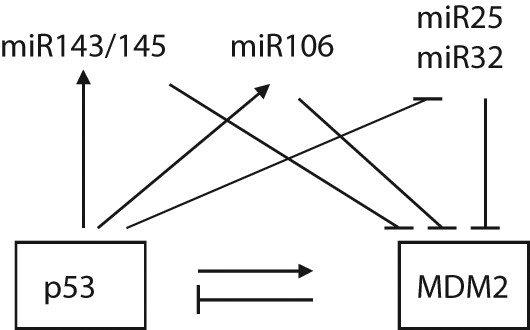
The regulation of MDM2 by microRNAs. A group of microRNAs, including miR143/145, miR605, miR23, and miR32, can down-regulate MDM2 through direct binding to 3′-UTR of the MDM2 mRNA, which in turn increases p53 protein levels and function. These microRNAs are either positively or negatively regulated by p53.
Conclusion and Future Perspective
As reviewed here, the p53 protein is activated and functions in response to a large and diverse set of stress signals. In addition to the stresses described here, the p53 pathway response is activated by telomere erosion (through the ATM pathway), hypoxia, heat and cold shock, reprogramming of epigenetic marks and stem cell formation, and denaturation of proteins in the cell. Remarkably, each stress signal has its own detector and pathway, but every one of these pathways funnels into a common node in the network, the MDM2 protein. The positive or negative regulation of the MDM2 protein ubiquitin ligase activity in turn regulates the p53 levels, and further protein modifications of the p53 protein activate it for transcription of selected genes. This results in a cellular response, including cell cycle arrest, DNA repair, apoptosis, or senescence, which in turn depends upon the stress, the cell type, the state of cancerous transformation, and the developmental time or stage of the cells and organism. By having p53 activity dependent upon the half-life of the p53 protein (which under nonstressed conditions is 6 to 20 minutes in different cells), this level of regulation is rapid with a protein concentration doubling every 6 to 20 minutes and protein modifications in seconds. A slower step comes in rolling out the transcriptional program of the p53 pathway. The organization of these stress response pathways brings up a number of interesting questions.
Why do so many diverse stress responses get funneled into a single node with MDM2 and p53 at the center? Mutations in the mdm2 gene (amplifications) or the p53 gene result in a high level of mistakes and mutations in cells that then develop into cancers. Why is this pathway not backed up with redundant activities? Is there an advantage of a single node that can then integrate diverse sets of stress signals (more than one stress at the same time)? Clearly, different pathways with diverse signals for MDM2 and p53 (sites of phosphorylation, acetylation, methylation, etc.) can carry a code that modulates different responses to stress. Where did this organization and uniform response to stress come from? What is the evolutionary path that the p53 family of genes has taken that results in protection from cancers over our lifetimes? p53 is one of a family of transcription factors along with p63 and p73. They share a great deal of homology in the DNA binding domains of the 3 proteins and even bind to the same DNA sequences. We can readily recognize p63/p73 ancestors in choanoflagellates, sea anemones, fruit flies, and roundworms 104 and p53 and MDM2 homologs in placozoans and spiders. 105 In these invertebrates, in every case where the functions of these gene products have been explored, the p53, p63, and p73 genes play a role in protecting the germline cells from DNA damage or starvation (glucose deprivation). Under these stress conditions, p53 family genes respond with cell death of germline cells, preventing developmental defects and monitoring the fidelity of the offspring. The p53 family proteins do this by using similar stress response pathways, activating a p53-like protein that binds to the same DNA sequences that the human p53 protein binds to and functions in a program of cell death. There is a preservation of structure and function over a billion years of evolution. In the vertebrates (as early as the cartilaginous fishes), the p53 protein moves from germline protection to somatic cell protection and cancer prevention. In humans and mice, all 3 of the p53/p63 and p73 gene products continue to play a role in the functions of the female germline and implantation of fertilized eggs. 106
In humans, what are the functions of the p53 protein? Is it more than protecting us from cancers? What are the stress signals that trigger cancers in humans which p53 prevents from happening? Is it oncogene activation via ARF, or is it DNA damage? Clearly, one function of the p53 gene is to protect the species from early-onset cancers. More than 50% of cancers harbor p53 mutations, and Li-Fraumeni syndrome patients harbor p53 germline mutations and develop cancers at an early age. High levels of p53 somatic mutations (60%-100%) are observed in BRCA-1 and triple-negative breast cancers and ovarian cancers, and these cancers are driven by DNA damage, so the p53 function in DNA damage control is an important protective response. While the ARF response to oncogene activation may be active in the formation of benign polyps in the colon (where APC mutations lead to high β-catenin levels that activate ARF and p53 controls polyp cell growth via apoptosis), this will require additional evidence to prove the point. The p53 protein can cause a number of pathologies while responding to stresses. Acute ischemia results in hypoxic death by p53 activation. Tissue damage is mediated by p53 death in the central nervous system (stroke) or heart. p53-null mice reduce this damage. 107 Similarly, radiation sickness and loss of the immune response with excessive radiation are a p53-driven response. The loss of eggs during chemotherapy is a p63 response, and lower levels of primary oocytes in an ovary are a p73-mediated event.108,109 Anorexia or glucose starvation results in oocyte death (likely a p63 event), lower estrogen levels, and failure to menstruate. The p53 family of genes is involved in inflammation and regulating the immune system. There will likely be an important role for the p53 gene family in stem cell regeneration and epigenetic changes. 110 Stem cell regulation in the central nervous system and neurodegenerative diseases have been suggested to involve p53 and p73.111,112 There may well be a price to pay for the diverse roles of p53 in stem cells. Several experiments suggest that too active a p53 response can deplete stem cells, in particular in bone marrow stem cells. 113 This could help to explain the suggestions that too high a level of p53 shortens the longevity of an organism. 113
There are strong tissue and cell type specificities in the p53 pathway and responses. The spectrum of tissues that develop tumors observed in people with Li-Fraumeni syndrome differs from the tissue spectrum of somatic p53 mutations observed in most cancers. Gene amplifications of MDM2 and MDM4 show distinct tissue specificity. Just why these mutational distributions are tissue specific remains a real mystery. Does MDM2 or MDM4 have functions in addition to p53 regulation? There are suggestions in favor of this idea, but this requires more studies. Are there other disorders, in addition to cancers, that p53 mutations can give rise to in the host organism? Have we examined in enough detail the metabolic pathways, immune system dysfunctions, central nervous system disorders, and reproductive disorders in individuals with Li-Fraumeni syndrome or in mice with different p53 mutant alleles? The p53 knockout mice have some metabolic deficits 114 with likely more to come.
As we begin to understand more about the pathways that connect physical and psychological stresses to the MDM2-p53 central node, we will likely find the answers to some of these questions. Just how we respond to stress, repair its consequences, and move back into a homeostatic state are key elements in reproduction free of errors or mistakes. The p53, p63, and p73 pathways enforce error-free reproduction by death to clones that harbor these mutations. As such, it limits variation both in the germline and the somatic tissue. In the germline, limiting variations slow the rates of evolution by narrowing the possibilities in the diversity of offspring. There is therefore a tension between an optimal rate of change and fidelity, and p53, p63, and p73 genes have evolved to function within this optimal window of protecting us long enough to reproduce ourselves and permitting enough mistakes so that natural selection can act upon a diverse genetic species of organisms.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: W.H. is supported by grants from the National Institutes of Health (1P30CA147892-01), Department of Defense (W81XWH-10-1-0435), the Cancer Institute of New Jersey (New Investigator Award), and the Ellison Foundation. Z.F. is supported by the National Institutes of Health (grant 1R01CA143204-01) and the New Jersey Commission on Cancer Research.