
Abstract
The transcription factor p53 regulates numerous cellular processes to guard against tumorigenesis. Cell-cycle inhibition, apoptosis, and autophagy are all regulated by p53 in a cell- and context-specific manner, underscoring the need for p53 activity to be kept low in most circumstances. p53 is kept in check primarily through its regulated ubiquitination and degradation by a number of different factors, whose contributions may reflect complex context-specific needs to restrain p53 activity. Chief among these E3 ubiquitin ligases in p53 homeostasis is the ubiquitously expressed proto-oncogene MDM2, whose loss renders vertebrates unable to limit p53 activity, resulting in early embryonic lethality. MDM2 has been validated as a critical, universal E3 ubiquitin ligase for p53 in numerous tissues and organisms to date, but additional E3 ligases have also been identified for p53 whose contribution to p53 activity is unclear. In this review, we summarize the recent advances in our knowledge regarding how p53 activity is apparently controlled by a multitude of ubiquitin ligases beyond MDM2.
Keywords
Functional Ubiquity in Ubiquitin Signaling
The complex orchestration of biological activities that allows eukaryotic life to exist and adapt to stressful environments depends exquisitely upon the tightly regulated abundance and activity of cellular proteins. The evolution of the ubiquitin-proteasome system is likely one of nature’s many responses to such environmental challenges. This mechanism of regulated protein turnover depends upon the sequential covalent attachment of multiple ubiquitin molecules to one or more target lysines of a substrate, followed by its recognition and proteolytic destruction by the multi-subunit 26S proteasome. This elegant system allows for the specific and rapid adenosine triphosphate (ATP)–dependent alteration of protein half-life and, therefore, transcription-independent temporal control of protein level and activity within the cell. Conjugation of ubiquitin to a substrate requires the concerted activity of a number of enzymatic activities. Ubiquitin is first activated by its ATP-dependent attachment to the ubiquitin-activating enzyme E1 via a thioester bond. This activated ubiquitin is then transferred to a member of the E2 ubiquitin-conjugating enzyme family, of which eukaryotic genomes encode a few dozen. A member of the E3 ubiquitin ligase family of proteins, comprising over 100 known factors, then catalyzes the processive transfer of multiple ubiquitin molecules to a substrate, either directly from the E2 to the substrate or with the E3 itself serving as a ubiquitin-transfer intermediate. A chain in which at least 4 sequential ubiquitins are linked through isopeptide bonds between lysine 48 and glycine 76 of a subsequent ubiquitin serves as the minimal substrate for recognition and subsequent destruction of the conjugated protein by the proteasome. However, in addition to these K48-linked ubiquitin chains, a number of alternate linkages (most notably through lysine 63) have also been described whose functions are independent of signaling proteasome degradation.1-4,5,6
Substrate specificity is primarily conferred through interaction of E3 enzymes or associated cofactors with unique sets of substrates; regulation of substrate ubiquitination is, therefore, largely controlled through the activity of E3 ubiquitin ligases. 7 Although many substrates are regulated by a single E3 ligase, in some cases, a substrate can be targeted for ubiquitination by multiple E3s to provide either redundancy or regulatory control specific to certain environmental or tissue contexts.8,9 This additional layer of complexity has potential implications for therapeutic targeting, a topic discussed thoroughly in a recent review. 10
In addition to the role of ubiquitination in protein degradation, a mounting body of evidence also indicates that regulated substrate ubiquitination also serves a number of critical signaling functions in the cell, many of which play important roles in tumorigenesis.11,12 Monoubiquitination and K63-linked polyubiquitination, signals insufficient to direct proteasomal degradation, have been shown in a number of experimental systems to affect receptor-ligand internalization and endocytosis, 13 protein conformation, 14 DNA repair,15,16 and inflammatory signaling.17-20 In addition, some evidence suggests that K48-linked ubiquitin chains, long assumed to exclusively signal a destructive fate mediated by the proteasome, can promote intramolecular substrate interactions that may mask recognition by the proteasome.21,22
Although energetically costly, regulation of protein abundance and activity through the ubiquitin-proteasome system permits rapid reduction of the levels and activity of those proteins whose functions must be shorter-lived than the protein’s unencumbered stability might otherwise allow. The products of tumor suppressor genes, whose activities are strongly selected against during tumorigenesis, represent such a class of proteins. Despite roles in promoting genomic integrity and proper cell-cycle regulation, tumor suppressor protein activities must be tightly controlled throughout an organism’s life span to circumvent deleterious pathological effects.
p53 is Controlled by Regulated Ubiquitination and Degradation
The fortuitous coupling of tumor suppression pathways with the ubiquitin-proteasome system during eukaryotic evolution has resulted in many examples of such critical restraint systems. Activation of the tumor suppressor p53, whose functional inactivation appears to be strongly selected for in virtually all cancers, is tightly associated with the control of its stability through regulated ubiquitination and degradation. Under normal conditions of low oncogenic and/or genotoxic stress, p53 is constitutively transcribed and translated, but its levels and activity are kept in check through continuous ubiquitination and proteasomal degradation. In response to these stresses, however, p53 ubiquitination and/or degradation is inhibited resulting in its rapid stabilization and activation.23,24 Since the identification of p53 as a substrate of the ubiquitin-proteasome degradation system, it has become clear that both p53 activity and stability are regulated, in some cases independently, by ubiquitination in nearly every facet of its physiologic response to stress. To date, over a dozen E3 ubiquitin ligases and other modulators of p53 ubiquitination have been identified through various approaches (Table 1). The relative contribution of each of these factors to p53 homeostasis and the p53 stress response will be the primary topic of in this review.
E3 Ubiquitin Ligases Reported to Directly Target p53 for Ubiquitination, Their Described Impact on p53 Activity, and Level of Validation
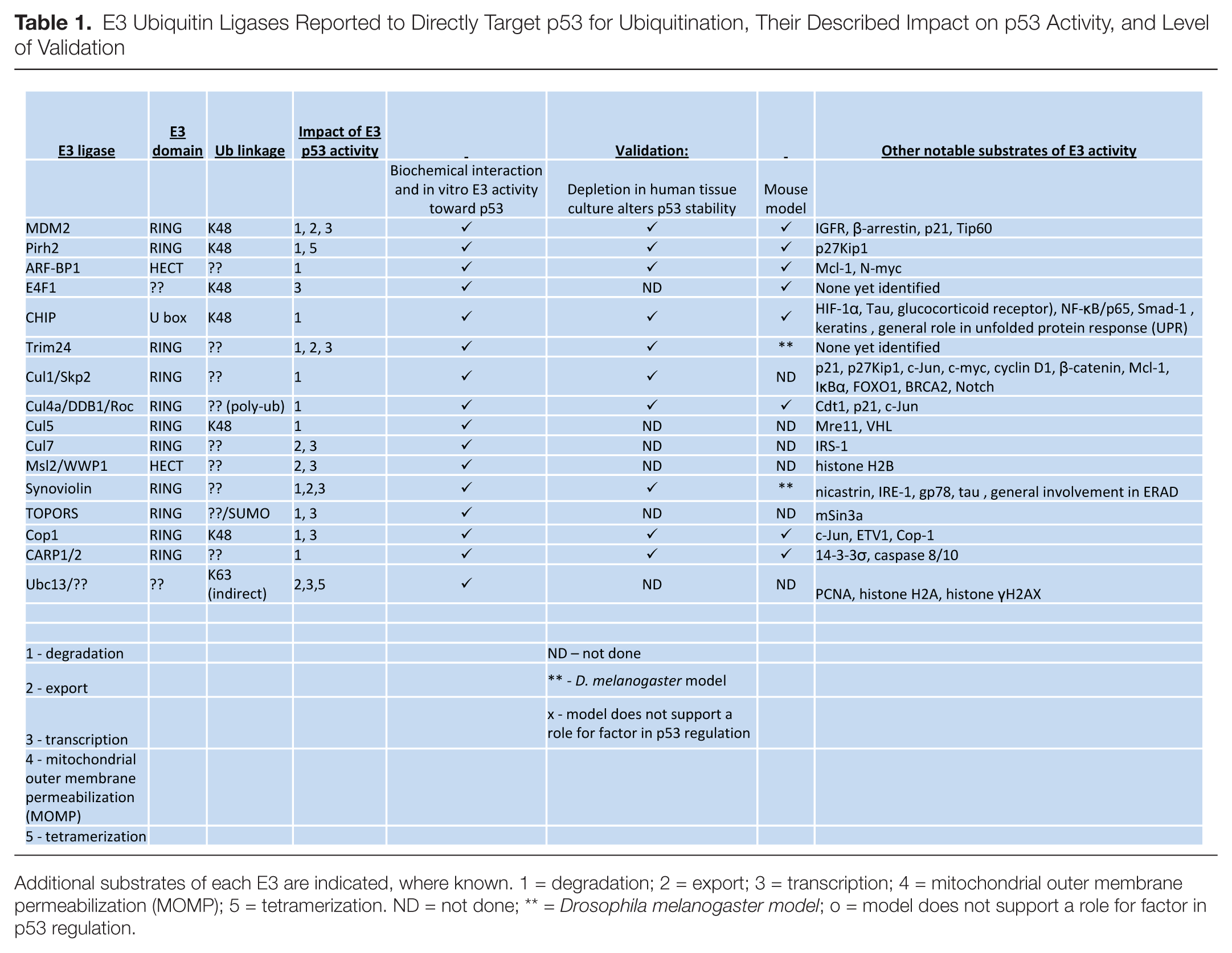
Additional substrates of each E3 are indicated, where known. 1 = degradation; 2 = export; 3 = transcription; 4 = mitochondrial outer membrane permeabilization (MOMP); 5 = tetramerization. ND = not done; ** = Drosophila melanogaster model; o = model does not support a role for factor in p53 regulation.
The Growing Family of p53 E3 Ubiquitin Ligases
Following initial reports that p53 is targeted for ubiquitin/proteasome-dependent degradation, the cellular E3 ligase E6AP, acting in concert with the high-risk HPV16 E6 protein, was identified as the first ubiquitin ligase responsible for promoting p53 degradation. 25 However, it quickly became clear that E6AP is competent for p53 degradation only in the context of high-risk HPV infection,26,27 raising the question of the factor(s) regulating p53 ubiquitination under physiologic conditions. In this section, the biochemical evidence supporting each factor as a putative p53 E3 ligase will be reviewed. The main focus will be on those factors that have been physiologically validated in cell and mouse models, but others that require further validation will be more briefly discussed, as a scarcity of physiologic data on an organismal level prevents integration of these ligases into the emerging and already incredibly complex network of validated p53-targeted E3s. This survey will be followed by a more thorough analysis of the contribution of each of these factors on an organismal scale, with a particular focus on human tumor etiology and experimental mouse models.
MDM2
During the time that E6AP-mediated p53 degradation was under intense investigation, another negative regulator of p53 function and stability was identified— the product of the mouse double-minute 2 gene (MDM2).28-30 MDM2 was first described as an inhibitor of p53-dependent transactivation through its ability to directly bind to and occlude the N-terminal p53 transactivation domain.30,31 It was quickly noted by a number of labs that the Mdm2 gene is itself a direct transcriptional target of p53,32,33 providing a plausible mechanism through which p53 activity could be reset to basal levels after induction by stress. Cementing its status as a critical negative regulator of p53, two simultaneous reports from the Bradley and Lozano groups indicated that mice lacking the Mdm2 gene die during early stages of embryonic development, but that concomitant deletion of p53 results in viable, albeit tumor-prone, mice born at expected Mendelian frequency.34,35 It was then shown in human and mouse tissue culture systems by the Oren and Vousden groups that MDM2 promotes p53 degradation in a manner dependent upon both MDM2/p53 interaction and functional proteasomes.36,37
Although providing convincing evidence of p53 antagonism by MDM2, these initial reports characterizing MDM2 destabilization of p53 did not address the detailed mechanism of this inhibitory activity and the specific role of MDM2. However, a report soon followed from the Yasuda lab demonstrating that MDM2 encodes an intrinsic, p53-directed E3 ubiquitin ligase activity. 38 It was subsequently shown that MDM2-p53 complexes serve to curtail p53 activity through continuous ubiquitination in unstressed cells and that upon UV or other genotoxic damage MDM2-mediated ubiquitination and degradation of p53 is disrupted, leading to p53 stabilization.39-41 Furthermore, analyses of MDM2 and p53 levels in irradiated cells indicated that MDM2 induction closely coincides with the postdamage return of p53 levels to basal values. 36 Together, the data accumulated during this short time from cell culture, biochemical, and mouse systems promote a model of p53 activation in which p53 is stabilized and activated through interruption of the p53-MDM2 complex and that the p53 response is rapidly dampened following p53-dependent induction of MDM2 expression. Subsequent experiments have convincingly supported and expanded upon this hypothesis, indicating that both disruption of the MDM2/p53 complex, inactivation of intrinsic MDM2 E3 activity, and transient destabilization of MDM2 by an ATM/ATR-initiated kinase cascade are necessary for the full physiologic stabilization and activation of p53 activity post-DNA damage.42,43
The biochemical p53-MDM2 interaction responsible for driving many of the negative regulatory functions of MDM2 toward p53 is highly complex and heavily regulated, indicating that the simple and commonly cited model described above provides an incomplete picture. Early evidence indicated a robust interaction between the MDM2 N-terminus and the N-terminal transactivation domain of p53,31,44 but more recent reports describe physiologically relevant interactions between MDM2 and the p53 C-terminus, 45 as well as the MDM2 acidic domain and the p53 DNA-binding domain, 46 the latter of which appears to be necessary for efficient ubiquitination of p53 by MDM2. 47 In vitro and cell-based interaction assays suggest that a complex array of posttranslational modifications and the tetramerization and DNA-binding state of p53 influence these interactions,48,49 although this knowledge has yet to be integrated into a complete model explaining precisely how p53 activity is regulated by MDM2. A number of mouse models have also since contributed greatly to our understanding of MDM2 in development and tumorigenesis, and the importance of these MDM2 model systems in delineating a unifying hypothesis of p53 ubiquitination will be discussed further below.
Pirh2
Subsequent to MDM2, the first E3 ligase directly shown to target p53 was another p53-induced RING domain E3, appropriately named “p53-induced protein with RING-H2 domain,” or Pirh2. 50 Leng et al. 50 endeavored to identify novel regulators of p53 ubiquitination through a differential display approach, contrasting mRNA expression of a mouse cell line harboring a temperature-sensitive p53 allele at either permissive or restrictive temperatures. The group identified and characterized Pirh2 as a p53-responsive, p53-interacting E3 ligase and, through overexpression and antisense experiments in immortalized human cells, showed that Pirh2 regulates p53 levels and transcriptional activity in a manner dependent upon its RING domain. Importantly, Pirh2 levels increased following irradiation in mouse embryo fibroblasts (MEFs) or cisplatin treatment in BJT fibroblasts, both in a p53-dependent manner. Other reports suggest that Pirh2 may only be induced by p53 in certain contexts. Stabilization and activation of wild-type p53 in human cancer cells (A549 and MCF-7) by γ-irradiation or cisplatin resulted in accumulation of MDM2 but not Pirh2. 51 Comparison of Pirh2 and MDM2-mediated p53 ubiquitination patterns 52 via mass spectrometric analysis of ubiquitinated p53 generated in vitro also indicates unique p53 lysine specificities for Pirh2 as compared to MDM2, with Pirh2 showing a preference as compared to MDM2 for several DNA-binding domain lysines, including K101, K162, K292, and K305. Notably, the lysine cluster at K319-K321 was not targeted at all by Pirh2 under these conditions, while they were subject to modification by MDM2. In contrast, the preference of Pirh2 or MDM2 for the C-terminal lysines of p53 did not appear to differ appreciably. Stoichiometric comparison of Pirh2 or MDM2 E3 activity toward p53 in vitro also suggests that Pirh2 bears weaker E3 ligase activity toward p53 than does MDM2, although in the absence of other regulators of ubiquitination, this may or may not reflect the case in vivo. Further highlighting this contrast, Pirh2 also exhibits a strong preference toward p53 tetramers, showing little or no E3 activity toward monomeric or dimeric p53 in vitro. 53
Further experiments have provided other intriguing hints that Pirh2 is involved in homeostatic regulation of p53. In human and mouse lung tumors overexpressing Pirh2 (93% and 84% of each tumor type, respectively), p53 ubiquitination was correspondingly increased. No detectable change in MDM2 transcript levels was observed in this study, although MDM2 protein levels were not examined. 54 Furthermore, phosphorylation of Pirh2 by CAM Kinase II, a calmodulin responsive kinase downregulated in a variety of tumor types, 55 reduces Pirh2 stability and impairs p53 ubiquitination in cells. 56 Pirh2 phosphorylation appeared to be regulated in a cell-cycle–dependent manner—CAMK II activity and Pirh2 phosphorylation peaked during G2/M, whereas p53 ubiquitination increased during the G1/S transition. Perhaps the strongest evidence supporting the role of Pirh2 as a physiologic E3 ligase for p53 comes from the recent characterization of Pirh2 –/– mice. 57 In contrast to MDM2, Pirh2 is dispensable for proper development in mice. However, in response to irradiation, splenocytes and thymii of Pirh2 –/– mice exhibited elevated levels of p53 and its targets p21 and PUMA, with this enhanced p53 activity resulting in increased apoptosis relative to wild-type littermates. Although a number of alternative explanations exist for this observation that require testing, the genetic and biochemical evidence, taken together, suggests that Pirh2 may play an important role in the sensitivity and potency of p53-dependent stress responses, and may regulate p53 in a cell-type specific manner.
ARF-BP1/MULE
The ARF tumor suppressor, encoded by the INK4A locus, is upregulated in the context of improper activation of several oncogenes and activates p53 by inhibiting MDM2 activity through mechanisms that may involve proteasomal degradation and nucleolar sequestration.58-62 In addition, however, ARF has also been shown to possess p53-independent roles in growth arrest. In an effort to identify ARF-interacting proteins involved in these processes, 63 researchers purified ARF complexes from p53-null cells and identified an ARF-interacting HECT-domain E3, which they named ARF-BP1. The E3 activity of ARF-BP1 was antagonized by ARF, and depletion of ARF-BP1 induced p53-independent growth arrest. Surprisingly, though, ARF-BP1 was also shown to regulate p53 stability and activity in several human cell lines expressing wild-type p53, as well as p53-transfected p53/MDM2-null MEFs. Subsequent in vitro and in vivo ubiquitin ligase assays confirmed p53 as a direct target of ARF-BP1 E3 ligase activity. Recently, ARF-BP1 was identified as a substrate of the ubiquitin-specific protease USP4. USP4-null mice exhibited decreased ARF-BP1 ubiquitination and stability, which correlated with increased p53 activity in mice splenocytes and thymocytes in response to ionizing radiation. 64
At the same time ARF-BP1 was identified as a ubiquitin ligase for p53, it was implicated as an E3 and functional antagonist of the oncogenic antiapoptotic factor Mcl-1. 65 Animal models accordingly paint a more nuanced picture of p53 regulation by ARF-BP1. In Caenorhabditis elegans, the ARF-BP1 homolog EEL-1 is necessary for germline cell apoptosis in response to genotoxic stress but does not appear to be involved in developmental apoptosis in somatic tissue. 66 In contrast, ARF-BP1 knockout in mice results in widespread b-islet p53 expression and apoptosis, culminating in early embryonic lethality. 67 To further explore physiologic functions of ARF-BP1 in tumorigenesis, the authors generated conditional knockout mice in which ARF-BP1 was deleted in pancreatic β-islet cells. These mice exhibited increased β-islet p53 levels, a reduced life span, severe diabetic symptoms, and progressive loss of glucagon and insulin staining in the pancreas. Strikingly, concomitant deletion of p53 in β-islet cells largely reversed both the diabetic and aging phenotypes, providing strong evidence that ARF-BP1 acts as a negative regulator of p53 in a physiologic setting.
E4F1
The transcriptional repressor E4F1 was initially described as an adenovirus E1A-regulated transcriptional repressor of adenovirus transcription, and was later shown to enhance p53-dependent cell-cycle arrest. 68 E4F1 has also been shown to regulate the activity of the hematopoietic stem cell (HSC) maintenance factor Bmi1, 69 whose inactivation in mice causes premature senescence associated with high levels of p53 and p19ARF. Le Cam et al. 70 subsequently demonstrated that E4F1 encodes an intrinsic p53-directed E3 ligase activity. Notably, E4F1 appears to bear no significant homology to classical HECT or RING domains (although it does encode a Zn-binding domain) and specifically targets lysines 319-321 of p53 for K48-linked ubiquitination. In striking contrast to other E3 ligases of p53, E4F1-mediated ubiquitination of p53 enhances p53 cell-cycle arrest activities but does not affect p53 localization or stability. E4F1 knockdown also sensitizes cells to both UV and ARF-induced p53-dependent apoptosis. These data suggest a role for E4F1 in the regulation in the regulation of cell-fate decisions downstream of p53 stabilization.
E4F1 deletion in mice results in embryonic lethality associated with a host of mitotic defects and apoptosis, although it is unclear whether the apoptosis reflects any contribution of spontaneous p53 activation or is a consequence of widespread mitotic checkpoint activation. 71 A mouse model in which E4F1 can be conditionally inactivated in the whole skin or basal epidermal layer by exposure to hydroxy-tamoxifen (4-OHT) 72 has provided some additional clues to the role of E4F1 in p53 regulation. Shortly after E4F1 inactivation in the skin by topical 4-OHT exposure, mice exhibit epidermal hyperplasia and hyperkeratosis, followed later by severe skin lesions, a loss of epidermal cellularity, and impaired wound healing. The authors found no evidence of p53-associated pathology such as increased apoptosis in E4F1-deficient skin. Further experiments indicated that the observed hyperplasia occurred as a result of improper regulation of proliferation and differentiation of the hair follicle (HF) stem cell compartment, with the resulting pathology due to exhaustion of the stem cell pool in E4F1-deficient skin.
The authors went on to show that deletion of the INK4A/ARF locus or depletion of p53 by shRNA partially restored clonogenic stem cell potential of E4F1-deficient HF stem cells. Overexpression of Bmi1, implicated in promoting stem cell identity and proliferation, also partially rescued this defect. Although these experiments do not provide direct evidence that E4F1 acts as a positive regulatory p53 ubiquitin ligase in vivo, the epidermal defects caused by E4F1 inactivation appear to be associated with increased p53 activity in stem cells. Unfortunately, a simple analysis of E4F1’s role in p53 homeostasis is complicated by the poorly understood interaction between E4F1, Bmi1, and the ARF-p53 pathway. Identification of the minimal E3 ligase domain of E4F1 and knock-in of a ubiquitin-ligase dead allele into mice could provide a useful tool to assess the importance of E4F1 E3 activity in its various physiologic roles.
Trim24
Recently, Allton et al. 73 undertook a novel approach to identify p53 binding partners. Reasoning that most p53-interacting proteins have been identified in transformed culture systems in which p53 regulatory controls are likely altered, the group engineered a tandem affinity purification (TAP) epitope into a single endogenous mouse p53 allele, allowing a high-stringency purification of p53 complexes likely enriched for relevant binding partners. The group isolated embryonic stem (ES) cells from the resulting mice and performed a mass spectrometric analysis of tandem affinity–purified p53 obtained from ES cell lysates. This analysis yielded MDM2, p53, the p53-binding partner 53BP1, and a number of novel p53-binding partners. The authors focused on one—the tripartite-motif factor Trim24—due to its overexpression in several tumors and its modulation during tumorigenesis by oncogenes such as Braf and Ret.74,75
Members of the Trim family contain canonical zinc-coordinating RING domains, although it is presently unclear whether all family members exhibit ubiquitin ligase activity. 76 Depletion of Trim24 by shRNA in mouse embryonic stem (ES) cells resulted in increased p53 levels, as well as activation of its transcriptional targets MDM2 and p21.73 Enhanced p53-dependent apoptosis in response to doxorubicin was also observed. The authors went on to show a robust interaction between p53 and exogenous Trim24 in several experimental systems. Importantly, these observations were recapitulated in several human cell culture systems. Cycloheximide analysis of p53 half-life in Trim24-overexpressing cells indicated that p53 half-life is reduced in the presence of exogenous Trim24 also exhibited an intrinsic E3 activity toward p53 in vitro. Notably, Trim24 appeared to competently polyubiquitinate p53, although whether this activity is present under physiologic conditions is unclear.
These experiments establish Trim24 as a probable regulator of p53 ubiquitination and degradation in vivo, but another innovative experiment in the same report implies that this critical role for Trim24 in keeping p53 in check is evolutionarily conserved. The authors chose to use Drosophila melanogaster as a model system to evaluate the importance of Trim24 on an organismal scale, as Drosophila encode a p53 homolog, yet no known orthologous MDM2 gene. Mosaic analyses of wing discs expressing mutant Bonus, the single homolog of the tripartite-motif family in Drosophila, demonstrated widespread p53-dependent apoptosis that was rescued nearly completely after dp53 RNAi-mediated depletion.
A follow-up report from the same group 77 indicates that the Trim24-p53 complex is disrupted during retinoic acid–induced human embryonic stem cell (hESC) differentiation. This suggests that one function of Trim24 in limiting p53 activity could relate to maintenance and proper differentiation of the hESC pool. Addressing these intriguing possibilities requires further analysis of the role of Trim24 in p53 regulation, stem cell maintenance, and tumorigenesis.
CHIP
Much of the focus in characterizing the network of negative regulatory factors targeting p53 has been related to mechanisms that blunt p53 activity during tumor development; however, a mutant allele of p53 is expressed in about half of all solid tumors and contributes to malignant progression through numerous gain-of-function activities.78,79 Most p53 mutant proteins exhibit strikingly increased stability relative to wild-type p53 in tumor cells, an effect long assumed to be due to the inability of mutant p53 to induce MDM2. However, growing evidence supports the notion that wild-type and mutant p53 ubiquitination and degradation are regulated differently; for example, mutant forms of p53 can be ubiquitinated, albeit in a largely MDM2-independent manner.80-82 Furthermore, mice engineered to express mutant p53 exhibit increased p53 stability only in tumor tissue, 80 strongly arguing that other E3 ubiquitin ligases contribute to mutant p53 ubiquitination and stability in vivo.
The E3 ligase C-terminus of HSP70-interacting protein (CHIP) is a prime candidate for this activity, as mutant p53 is often found in HSP70 and HSP90 chaperone complexes.83-86 With the exception of MDM2, CHIP is perhaps the most well-characterized ubiquitin ligase believed to target p53. The primary function of CHIP appears to be promoting the ubiquitination and degradation of misfolded proteins.87-89 CHIP has also been implicated in the clearance of protein aggregates common to many neurodegenerative disorders, although the involvement of CHIP in the underlying pathogenesis of these diseases is extremely complex and poorly understood.
A role for CHIP in p53 ubiquitination arose from the observation that CHIP interacts with mutant, but not wild-type p53, and that CHIP overexpression in p53-null H1299 cells dramatically reduces mutant p53 levels and half-life. 90 Surprisingly, CHIP also appears to reduce wild-type p53 levels in this system. CHIP was also confirmed to function as a p53 ubiquitin ligase in vitro, and CHIP knockdown in U2OS cells, which harbor wild-type p53, appears to spontaneously increase p53 levels and activity.
Several other reports support an important role for CHIP in p53 activity. Recent work suggests that CHIP enhances p53 DNA binding through its activity as a chaperone by promoting the proper folding of p53 during heat stress 91 and that CHIP may be the primary E3 ligase for mutant p53.81,92,93 CHIP-mediated degradation of mutant p53 is enhanced in cancer cells in which HSP90 is inactivated pharmacologically by RNA interference 84 or by the HDAC inhibitor SAHA. 94 The authors suggest that Hsp90 inactivation frees mutant p53 from a protective HSP90 chaperone complex, sensitizing it to CHIP and MDM2-mediated ubiquitination.
Some recent evidence also suggests an important role for CHIP in controlling wild-type p53 activity. p53 has previously been described as a major contributor to heart failure under conditions of hypoxia,95-97 prompting the authors of one recent report to identify negative regulators of p53 expressed in heart tissue. Informing upon the potential physiologic role of CHIP in p53 regulation, CHIP was identified in a human heart cDNA expression screen as a suppressor of p53 transactivation using a p53 transcription reporter assay. 98 Depletion of CHIP in cardiomyocytes elevated p53 levels, and CHIP heterozygous mice also displayed elevated p53 levels relative to wild-type mice. Overexpression or pharmacological activation of CHIP in the heart protected mice from ischemic injury and strikingly reduced apoptosis in cardiomyocytes. Importantly, the increase in cardiomyocyte p53 levels after ischemic injury was greatly blunted in both settings, suggesting that CHIP plays a physiologic role in regulating cardiac responses to hypoxia through p53.
A CHIP-null mouse model has also been generated which is characterized by perinatal lethality (approximately 20% of CHIP –/– mice), likely due to excessive thymic apoptosis. MEFs of surviving CHIP-null mice exhibited increased sensitivity to heat shock–induced apoptosis, a stress response known to result in p53 stabilization in both wild-type and mutant p53-expressing cells.99-103 Although the apoptosis observed in these experiments may reflect p53-dependent caspase activation, it is likely a physiologically triggered heat shock response rather than a failure of homeostatic p53 regulation, as most CHIP-null mice appeared phenotypically normal unless exposed to heat stress. This critical role for CHIP in regulating the heat shock response makes dissecting its role in the regulation of p53 stability a challenge.
Still, a physiologic role for CHIP in mutant p53 regulation may stand to reason in light of recent work reporting the aggregation of mutant p53 with a number of tumor suppressors, including p63 and p73. 78 In this context, p53 aggregates may indeed be cleared by ubiquitination and degradation promoted by CHIP, although the known interplay between p53 and other heat shock response proteins,84,94,104-108 such as Hsp70 and Hsp90, complicates a simple hypothesis describing how p53 might be regulated by the heat shock response system in unique conditions of physiologic stress. Even so, we believe that the mounting evidence implicating CHIP in p53 stability control necessitates that it be considered in models concerning p53 homeostasis.
Cullin Complexes (Cul4a, Cullin 7)
The 7 known human cullin proteins serve as scaffolds for the assembly of several heteromultimeric E3 ligase complexes, all of which are believed to be activated upon conjugation of the ubiquitin-like modifier NEDD8, and work in conjunction with the COP9 signalosome (reviewed in Lee and Zhou 109 ). These complexes are composed of several adaptor proteins and at least one E3 ligase, ROC1. Cullins are often deregulated during tumorigenesis, and numerous reports of p53 antagonism by various cullin complexes exist. To date, roles for Cul1,110,111 Cul4a,112-115 Cul5, 116 and Cul7 117 in modulating p53 activity have been described, as well as cullin complex adaptor subunits DDB1, 118 Skp1,110,111 JFK, 110 and Skp2. 119
In addition, biochemical evidence exists that Cul1, Cul4a, Cul5, and Cul7 complexes directly ubiquitinate p53. Cul5 is necessary for p53 ubiquitination by adenovirus factors E4orf6 and E1B55k, 116 although to date no adenoviral protein-independent physiologic role for Cul5 complexes in p53 ubiquitination has been described. Inactivation of the Cul1-containing SCF complex by HIV-1 Vpu has been shown to inhibit p53 ubiquitination, leading to p53-dependent apoptosis. 111 Two reports indicate that Cul4a complexes promote MDM2-dependent p53 polyubiquitination and degradation—the first 114 describes an MDM2-Cul4a complex that participates in p53 degradation in human cells, delaying p53 accumulation after DNA damage in an MDM2-dependent manner. A subsequent account further characterizes the role of Cul4a in p53 ubiquitination by showing robust in vitro and in vivo E4 ligase activity toward p53; interestingly, this activity is dependent upon both MDM2 and the Cul4a complex E3 ROC1. 115 The authors also provide evidence that the Cul4a/DDB1/ROC1 complex may regulate MDM2 levels following DNA damage, an intriguing possibility that requires further study. In contrast to the other cullin complexes, Cul7-mediated ubiquitination of p53 appears largely MDM2 independent, promotes only monoubiquitination of p53 in vitro, does not appreciably alter p53 half-life in cells, and yet still antagonizes p53 activity following UV irradiation. Surprisingly, a ROC1-binding deficient Cul7 remains competent for in vitro p53 monoubiquitination, suggesting either an intrinsic Cul7 E3 ligase activity or the presence of another E3 in the Cul7 complex. 117
Genetic models in mice and C. elegans have been described to explore the roles of various cullins in p53 function during development and tumorigenesis. Gao et al. 120 uncovered a role in C. elegans for an evolutionarily conserved SCF complex in the suppression of germline apoptosis by the p53 homolog CEP-1. Inactivation or knockdown of Cul1, several Skp1-related genes, or the F-box protein FSN-1-resulted in increased germline apoptosis after DNA damage by the DNA-alkylating agent ENU. This effect was reversed by CEP-1 knockdown, suggesting that the observed apoptosis was CEP-1 dependent. It will be critical to determine whether germline regulation of p53 by cullin complexes is similarly regulated in mammals.
Mouse models addressing the role of DDB1 in development and genomic stability indicate that this member of the Cul4a complex is required for proper regulation of epidermal proliferation, as its deletion in the epidermis leads to widespread genomic instability and p53-dependent apoptosis. 121 Targeted inactivation of DDB1 in the brain resulted in a strikingly similar phenotype characterized by genomic instability and p53-dependent apoptosis. 118 MEFs containing floxed DDB1 alleles showed sensitivity to UV exposure and exhibited higher levels of p53 and target genes p21 and Bax after DDB1 deletion. 113 Complete DDB1 inactivation in the developing mouse embryo resulted in early embryonic lethality, suggesting that DDB1 and the Cul4a complex could be a ubiquitous regulator of the cell cycle.113,118 Concomitant deletion of p53 resulted in a partial rescue from cell death; however, whether this complex participates directly in the ubiquitination of p53 in vivo will require further analysis.
Other E3 Ligases of p53 Awaiting Full Validation
Although several other factors harboring E3 ligase activity toward p53 have yet to be fully validated in organismal systems, these factors merit mention in this review, as they may indeed play critical physiologic roles in the regulation of p53 function.
COP1 is a RING domain protein and member of the COP9 signalosome for which biochemical evidence supports an important role in p53 degradation. COP1 was identified as a binding partner of p53 through mass spectrometric analysis of FLAG-COP1–bound peptides eluted from U2OS cell lysates. 122 COP1 overexpression reduced p53 stability and activation of p21 in a RING-dependent manner, and its depletion resulted in stabilization of p53 and induction of p21. 122 Further evidence supporting COP1’s role in p53 homeostasis remains limited; however, a recent report implicates COP1 in a p53-regulatory feedback loop in human keratinocytes. 123 This is an especially exciting observation in light of several experiments suggesting that MDM2 is not induced by UVB-activated p53 in keratinocytes.124,125 Confounding the simple hypothesis that COP1 functions as an oncogenic E3 ligase for p53 was the observation that c-Jun is also targeted for degradation by COP1.126,127 A recent mouse model attempted to clarify the physiologic role of COP1 128 —while COP1 null and heterozygous mice die by E9.5 and E12.5, respectively, no signs of widespread apoptosis or decreased mitosis were evident to suggest altered p53 activity. To circumvent the embryonic lethality of COP1 –/+ mice, mice homozygous for a hypomorphic COP1 allele were generated to further probe its putative role in tumorigenesis. c-Jun was hyperactive in this model, whereas no elevation in p53 levels or activity was observed. In accordance with a critical role in blunting c-Jun proliferative activity, many COP1 hypomorphic mice developed spontaneous T-cell lymphoma. This surprising identification of COP1 as a tumor suppressor will require a more careful analysis of the p53 response in various tissues of COP1-deficient mice to define whether COP1 does indeed play any role in p53-mediated tumor suppression.
Although implicated in p53 signaling, a number of additional factors rely on limited biochemical evidence to support their putative E3 ligase activity toward p53 due to technical or other difficulties in generating a meaningful physiologic model. A full understanding of these E3 ligases, including Msl2/WWP1,129,130 CARP1/2,131,132 the endoplasmic reticulum resident protein synoviolin, 133 and the ubiquitin/SUMO E3 ligase TOPORS,134,135 merit mention but will require further study in physiologic systems to establish their validity as p53-directed E3 ligases.
Regulation of p53 Polyubiquitination by E4 Ubiquitin Ligases
Mounting evidence supports the notion that p53 is regulated at numerous points even after monoubiquitination by an E3. E4 ubiquitin ligases, which directly ubiquitinate monoubiquitinated substrates or serve scaffolding functions to increase E3 processivity, have been shown to play critical roles in p53 activity as well. MDM2 has been shown in a number of systems to catalyze monoubiquitination of substrate lysines in vitro but exhibits little or no polyubiquitin ligase activity at physiologic concentrations, 136 suggesting that an additional cofactor or ubiquitin ligase activity is necessary to promote efficient p53 polyubiquitination.
The importance of understanding the regulation of p53 mono- and polyubiquitination is highlighted by the starkly contrasting activities of mono- versus polyubiquitinated p53: Several studies support the notion that monoubiquitinated p53 is exported to the cytoplasm, where most polyubiquitination seems to occur.10,129,136 Nuclear export of monoubiquitinated p53 has been linked to mitochondrial localization of p53, where it directly participates in apoptosis by enhancing mitochondrial outer membrane permeabilization.137-140 In addition, cytoplasmic p53 has been linked to the inhibition of autophagy. 141 Polyubiquitination of p53 may therefore represent a critical regulatory step in p53-dependent cell-fate decisions through controlling the balance of p53 mitochondrial localization versus proteasomal degradation.
E4 ligases of p53 to date can be broadly classified in 2 groups: factors that bear intrinsic ubiquitin ligase activity toward monoubiquitinated p53 and those that most likely enhance the processivity of an E3142,143 or its interaction with p53. Members of the former group include, to date, the cullin 4a complex and the paralogous acetyltransferases p300 and CBP. p300 and CBP do not encode a conserved E3 ubiquitin ligase domain but bear intrinsic ubiquitin ligase activity distinct from their acetyltransferase domains10,144 and strongly enhance polyubiquitination of p53 in vitro and in vivo. A novel “two-step” assay using purified monoubiquitinated p53 as a substrate has demonstrated that p300 and CBP E4 activity acts directly on monoubiquitinated p53 in vitro. Efforts aimed at defining the biochemical nature of these novel E4 ubiquitin ligase activities, as well as their impact on p53 stability and activity, are ongoing.
Notable E4 ligases that enhance polyubiquitination of p53 by MDM2 include the E3/E4 UBE4B, 145 the proteasome-associated factor gankyrin, 146 and the transcription factor YY1. 147 UBE4B is the mammalian homolog of yeast UFD2, the first identified E4 ubiquitin ligase. 143 Each of these factors has been shown to promote p53 polyubiquitination in the presence of MDM2 in vitro and in cells, but precisely how these factors regulate p53 polyubiquitination may differ. YY1 appears to enhance p53 polyubiquitination primarily through enhancing the p53-MDM2 interaction. In contrast, gankyrin directly enhances MDM2 E3 ubiquitin ligase activity and the association of polyubiquitinated p53 with the proteasome. Although neither of these factors appears to possess an intrinsic ubiquitin ligase activity, UBE4B bears E3 activity encoded by a RING-like domain known as a U-box, the presence of which is necessary for polyubiquitination of p53 facilitated by UBE4B.
MDM2 itself appears to promote p53 polyubiquitination under certain conditions 136 when MDM2 is present in excess of p53. Although this observation holds true both in vitro and after overexpression in culture, it is unclear whether the MDM2:p53 ratio reaches a threshold under physiologic conditions where MDM2 itself drives p53 polyubiquitination. Still, these observations raise the intriguing possibility that under basal nonstress conditions, MDM2 drives monoubiquitination and nuclear export of p53 and relies on other E4 activities to direct polyubiquitination in the cytoplasm, while during p53-activating stresses, MDM2 reaches sufficient levels to promote p53 polyubiquitination in the nucleus. If true, such an additional level of p53 regulation would likely affect p53 localization and apoptotic potential, a possibility warranting further investigation.
Discussion
Why Do Multiple E3 Ubiquitin Ligases Target p53?
A signaling node as powerful as p53, bearing a potent ability to permanently remove cells from an organism’s replicative pool, must certainly be kept in check carefully to allow tumor-suppressive activities to be engaged while posing a minimal threat to organismal development and continued survival. For a number of reasons that may still not be fully appreciated, evolutionary processes have primarily selected a posttranscriptional method of proteasomal destruction to limit p53 activity through tight regulation of protein levels. In this light, the broad heterogeneity of genotoxic stresses and unique determinants of cell identity in various tissues make the simple expectation of a single E3 ligase in controlling p53’s multitude of complex tasks seem overly simplistic.
To date, over a dozen E3 ligases have been reported to target p53 (Table 1)—a figure well in line with the simple presupposition that temporal and tissue-specific honing of p53 responses may have conscripted a number of negative regulators during evolution. The type and extent of genomic stresses experienced by different tissues or during discrete developmental stages can differ strikingly, suggesting that selective processes could have selected for stress responses least detrimental to the organism. Responses of different tissues to genotoxic compounds or radiation do differ significantly throughout development and in at least a partially p53-dependent manner. 148 Developmental and differentiation defects in p53 mouse models have been observed, such as exencephaly in p53-null mice,149,150 and increased differentiation of mesenchymal kidney cells in p53 transgenic mice.151,152 These lines of evidence confirm that certain tissues and/or developmental stages possess different intrinsic sensitivities to alterations in p53 regulation that may greatly affect cellular responses to genotoxic stress, as well regulation of differentiation and proliferation.
Despite the years of painstaking efforts that have resulted in the identification of the numerous p53 E3 ligases described above, the question of how multiple factors might coordinate the multitude of p53 functions in the context of an organism remains a mystery. Although it is possible that a single E3 ligase regulates p53 activity and stability under physiologic conditions, there are a number of reasons that evolutionary processes may have selected several such factors to control p53 (Fig. 1). Increasing tissue complexity, cell number, and longevity of more complex organisms may all have necessitated constant reorganization of p53 regulation and signaling by ubiquitin ligases.
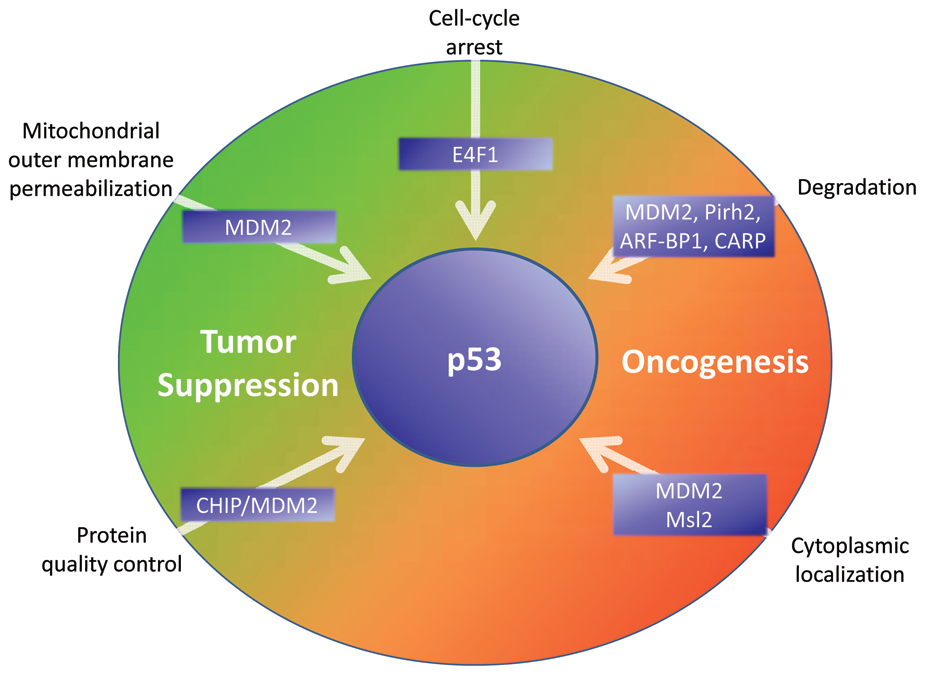
Impact of ubiquitination by specific E3 ligases on facets of p53 regulation and activity. Based on whether the E3 activity activates or inhibits a given p53 activity, the relationship of the indicated E3 activities to tumor suppressive or oncogenic effects is indicated.
For these reasons, it seems likely that metazoans, to avoid the negative consequences of unrestrained p53 activity, have encoded at least one potent negative regulator of p53 activity since its evolution. Indeed, the proapoptotic capacity of p53 is conserved at least to organisms as primitive as the sea anemone, 153 although how p53 activation is regulated in these organisms is not yet well understood. Although it is easy to surmise that any p53 E3 ligase conserved throughout metazoan evolution would retain its activity toward p53, numerous selective pressures could have promoted the actions of additional E3 ligases, possibly even eclipsing the function of an antecedent E3 ligase. Identification and characterization of E3 ligases in organisms representing a broad phylogeny may also provide much-needed insight into how the p53 circuit has been governed by E3 ligases during metazoan evolution.
What Current Mouse Models and Human Genetic Evidence Tells Us
Although traditional genetic mouse knockout systems have been critical in defining the roles of several ubiquitin ligases in regulation of p53, several recent conditional models have allowed a more nuanced approach in determining the contexts in which these ligases may regulate p53. Combined with human genetic and tumor studies, a comprehensive picture is beginning to arise with regard to p53 homeostasis in normal tissue and how these processes may be subverted during tumorigenesis. The most convincing evidence implicates Pirh2, MDM2, ARF-BP1, and CHIP in regulation of p53 ubiquitination. The potential importance of each of these factors in the control of p53 activity, as well as a few of the many remaining challenges in developing a holistic model of p53 function, will be described below.
Since the seminal discovery of MDM2 as a critical repressor of p53 activity in vivo,34,35 a number conditional mouse models have been generated to more finely probe the interplay between MDM2 and p53. Several approaches have been taken to sidestep the embryonic lethality of MDM2-null mice; hypomorphic MDM2 alleles, 154 MDM2 heterozygous mice, 155 temporal reactivation of p53,156,157 tissue-specific MDM2 knockout,158,159 knock-in of MDM2 point mutants, 157 and transgenic MDM2 overexpression160,161 have all greatly contributed to our understanding of the MDM2-p53 regulatory axis. Critical roles for MDM2 have been described in several tissue types—osteoblasts, 158 red blood cells, 162 lymphoid and hematopoietic lineages,154,163 and the epidermal stem cell compartment. 159
A few models merit special consideration here with regard to their impact on our understanding of p53 degradation in vivo. A recent mouse model developed by the Evan lab, 164 allowing spontaneous reactivation of p53 in which p53 is fused to the hormone-binding domain of estrogen receptor (ER), 165 has provided a wealth of information on the topic. Upon administration of the ER-ligand hydroxyl-tamoxifen (4-OHT), the p53-ER fusion (p53-ER) is rendered competent for activation. Despite a slight blunting of p53 target gene expression, life span, tumor incidence, and radiosensitivity of the p53-ER(TAM) mice are nearly identical to normal p53 +/+ mice, indicating that fusion of the modified ER domain has minimal, if any, impact on p53-dependent stress responses when tamoxifen is present. The Evan lab 156 used this p53-ER(TAM) system to reactivate p53 in MDM2-null, p53ER (TAM)/– mice to evaluate the impact of p53 reactivation in an MDM2-null background. Reactivation of p53 subjected these mice to widespread and spontaneous apoptosis in all tissues examined and was invariably lethal even after transient p53 restoration by a single dose of 4-OHT. Strikingly, despite a slightly higher half-life, p53-ER(TAM) was still efficiently degraded upon 4-OHT administration under nonstress conditions, providing evidence that alternative E3 ligases may cooperate in the ubiquitination and degradation of p53. These data strongly support the notion that, even after development, MDM2 is a critical suppressor of p53 activity, while still affirming that p53 is regulated by additional E3 ubiquitin ligases.
A subsequent report using the same p53-ER(TAM) system explored the impact of the MDM2 RING domain in p53 homeostasis in the adult mouse. The MDM2 point mutant 462A (464A in humans), deficient in ubiquitin ligase activity, was knocked into mice to determine the requirement of MDM2-mediated ubiquitination in regulating p53. 157 Surprisingly, the authors found that mice homozygous for Mdm2 C462A succumbed to p53-dependent embryonic lethality, as determined by the viability of C462A/C462A mice crossed with p53-null mice. To further study the impact of MDM2 lacking E3 activity, the authors turned to the p53-ER(TAM) system to circumvent the lethality of this point mutant knock-in. Their experiments strongly indicate that the 462A mutant retains full p53-binding capability, proving that the p53-MDM2 interaction is not sufficient for p53 suppression. The authors further showed that, upon release by 4-OHT, p53-ER(TAM) half-life is much higher in the 462A/462A mice as compared with Mdm2 +/+ mice but that MDM2 half-life itself is unaffected by loss of E3 activity. The reason for the observed discrepancy in p53 half-life between the two systems is unclear; while MDM2-null mice are able to target the p53-ER fusion for degradation upon genotoxic insult, MDM2 462A/462A mice are deficient in p53-ER degradation. This may be a consequence of the retention of p53 binding by MDM2 462A, which could occlude p53 docking sites of other E3 ligases, or of other E3-independent activities of MDM2. The data generated from these conditional mouse models convincingly indicate that the existing model of p53 regulation by MDM2 as the sole E3 for p53 requires revision.
CHIP may also play a role in p53 ubiquitination under certain conditions. Mounting evidence implicates the heat shock response in the regulation of p53 ubiquitination and stability through the assembly of Hsp70-p53 and Hsp90-p53 complexes. Many p53 mutants exhibit impaired thermodynamic stability relative to wild-type p53, perhaps suggesting roles for Hsp70 and Hsp90 in promoting proper folding of mutant p53. Indeed, a recent report indicates that the oncogenic gain of function of a subset of p53 mutants is attributable to its increased aggregation propensity. 78 Under conditions of heat shock stress or excessive protein translation, it is conceivable that wild-type p53 could also require chaperone functions to promote its folding and prevent aggregation with other nascent peptides. This possibility will need to be evaluated, possibly through the use of CHIP-conditional knockout or knock-in mice to allow the contribution of CHIP to p53 homeostasis under specific stress or nonstress conditions to be evaluated.
Limitations of Common Experimental Systems
Although the many genetic systems being used to dissect the role of p53 in tumorigenesis each harbor distinct advantages, great care must be taken in the interpretation of data obtained from these models for a number of reasons. The most obvious and commonly mentioned problems arising during data interpretation relate to de novo phenotypes generated as artifacts of protein overexpression. Transfection stress, altered stoichiometry of protein complexes, and forcing of nonphysiologic interactions by overexpression must all be considered in the evaluation of data obtained from these systems. Indeed, forced polyubiquitination or targeting of a substrate to the proteasome is sufficient to induce substrate degradation, 166 suggesting that artifactual, nonphysiologic interactions of p53 with many ubiquitin ligases could likely force its ubiquitination and/or degradation. Although overexpression is often a culprit in discrepancies between experimental systems, all model systems are characterized by distinct disadvantages that require consideration, especially in the study of a sensitive stress-responsive protein such as p53.
The culture of human cells provides a setting in which the contribution of many genetic and epigenetic differences to p53 function between humans and other species can be largely eliminated. However, it is critically important to consider the alien environment in which these cells proliferate—a landscape constitutively flooded with high levels of growth factors to which cells are rarely exposed in a true physiologic environment. Selective pressures unique to cell culture environments will certainly select for those clonal populations best suited to grow in these environments, resulting in numerous genetic and epigenetic alterations to cell growth and proliferation programs. In addition, many human culture systems are generated from tumor tissue, which has already selected against numerous genomic integrity, proliferative control, and apoptotic control mechanisms. These observations even raise the possibility of E3 ligases being selected to promote p53 degradation in culture, which are otherwise entirely irrelevant to p53 function under physiologic conditions. For these reasons, it may be beneficial to view human tissue culture systems as artificial experimental environments harboring human genomes, rather than cell populations truly representative of those constituting living organisms.
As a consequence of the inherent characteristics of various experimental systems, approaches aimed at defining E3 ubiquitin ligases that target p53 could result in the identification of factors that do not target p53 under physiologic conditions or, conversely, failure to identify an E3 that is physiologically relevant to p53 homeostasis. With p53 sitting at the center of a major stress node, it is conceivable that depletion or overexpression of many factors could affect a variety of physiologic stress responses to which p53 levels and ubiquitination are highly sensitive. Additionally, depletion or overexpression of an E3 likely to target dozens or more substrates for ubiquitination will necessarily affect the expression levels and activity of many other factors—in some cases, even other E3s. 167 Often, E3s target substrates harboring opposing activities in tumorigenesis,57,128 complicating the simple analysis of these E3 ligases in tumor suppression by p53. These possibilities highlight the need to validate target E3s in additional assays—probing of several physiologic p53-responsive stresses such as hypoxia, double-strand break (DSB) accumulation, and oncogenic stress to ensure they are minimally perturbed by modulation of a target E3, as well as thorough validation of p53 as a direct and robust target of an interrogated E3 via in vivo and in vitro ubiquitination assays.
The characterization of p53 ubiquitin ligases in mouse models presents additional challenges. E3s, which may not function as universal regulators of p53 levels, present one challenge—perturbation of such factors by genetic knockout or transgenic overexpression could elicit subtle or tissue-specific phenotypes that yield no gross anatomical or physiologic abnormalities. Compensation of gene knockout by functionally overlapping factors could also mask the identification of ubiquitin ligases targeting p53; however, the utility of therapeutically targeting such factors would likely be limited in this situation unless the compensatory factors were identified. Finally, E3s that target p53 only under very specific conditions may exist. With p53 playing a vital role in such disparate activities as DNA repair, 168 senescence, 169 apoptosis, 170 and mitochondrial respiration, 171 to name a few, the challenge will fall on the scientific community to continue developing innovative assays at the organismal level to further our understanding of p53 regulation by E3 ubiquitin ligases.
Organismal Differences
An added and often overlooked level of complexity in interpreting results from nonhuman genetic systems stems from the fact that organismal physiology has changed significantly throughout metazoan evolution, reflecting adaptation to different environments encompassing unique assortments of both intrinsic and extrinsic genotoxic or other environmental stress. Indeed, the evolution of the p53 transcriptional network has changed markedly even from rodents to primates. Specifically, roles for p53 in DNA metabolism appear unique to primates whereas several apoptotic genes regulated by p53 in mice do not appear to be p53-responsive in primates. 172 These observations raise the possibility that stress responses unique to certain evolutionary lineages may require unique p53 E3 ligases.
Indeed, many broad physiologic distinctions between mice and humans are likely to affect tumorigenic processes, which could have promoted concomitant adaptation of the p53 response. Most notably, and in contrast to primary human tissue, telomerase is active in most mouse tissues, highlighting a substantial difference in the regulation of a process well known to contribute to human tumorigenesis. 173 Also of critical importance to our full understanding of p53-mediated tumor suppression is the fact that ARF appears to be regulated differently between mice and humans—the potent response of mouse ARF to Ras overexpression in MEFs is not recapitulated in primary human fibroblasts,174,175 and ARF-null mice are severely tumor prone, 176 in contrast to humans, in which mutations in ARF that spare p16INK4A are relatively rare. 177 On an even more basic level, the stark contrast in cell number and lifespan between these species indicates that processes governing cell-cycle control, and therefore the lifetime extent of DNA replication-associated genotoxic stress, necessarily differ.
Future Perspective
Despite these shortcomings, probing of the p53 E3 ligase network will ultimately require the use of conditional mouse models, wherein the activity of p53 or its various ubiquitin ligases can be controlled in a temporal and/or tissue-specific manner to define contexts in which the activity of specific E3 ligases is necessary to control p53. Studies of the temporal development of tumors driven by oncogenes or environmental stresses will also be critical in supporting roles for p53 E3 ligases in tumorigenic processes. With these experimental systems, a broad range of physiologic stresses can be examined, the dependence of the ensuing p53 response on various combinations of regulatory factors measured by various “-omic” approaches, and a thorough and comprehensive model of p53 regulation by E3 ubiquitin ligases developed.
The biochemical basis of p53 ubiquitination has received much attention but is still not well understood. In addition, the interplay of p53 E3 ligases in a broader physiologic setting also remains mysterious. The need for such a multitude of factors that, excluding the function of the purely regulatory p53 E3 ligases, seem to direct the same outcome—p53 polyubiquitination and proteasomal degradation—presents a puzzling conundrum. It is possible that tissue specificity or developmental cues regulate unique E3 ligases under specific environmental contexts. Indeed, MDM2 itself is differentially modified in certain contexts by diverse factors such as AKT, 178 ATM,179-181 and GSK3, 182 leading to altered substrate specificity, ubiquitin ligase activity, and DNA damage response kinetics.
The bulk of current evidence supports a role for MDM2 in the physiologic regulation of p53 in many tissue types and both during development and in fully developed adults. We propose that additional E3 ligases such as Pirh2, CHIP, and ARF-BP1 exist for p53 that fine-tune the p53 response through modulation of its levels or activity by ubiquitination and that these E3 ligases are necessary for p53 regulation in specific tissue types, during specific developmental stages, or in response to unique types or combinations of physiologic stress (Fig. 1).
Footnotes
Acknowledgements
We thank S. Deb and S. P. Deb for critical review of the manuscript and helpful comments.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: SRG is supported by NCI R01-CA107532.