
Abstract
Tumor angiogenesis, the building of blood vessels in an expanding tumor mass, is an elegantly coordinated process that dictates tumor growth and progression. Stromal components of the tumor microenvironment, such as myofibroblasts and the extracellular matrix, collaborate with tumor cells in regulating development. Such myofibroblasts and the extracellular matrix have ever-expanding roles in the angiogenic process as well. This review summarizes how stromal myofibroblasts and the extracellular matrix can modulate tumor angiogenesis, highlighting recent findings.
Introduction
Angiogenesis, the process of building new blood vessels from existing ones, is a pivotal step in tumor development. The rate of tumor growth and progression relies on the tumor vasculature to provide a steady supply of nutrients and oxygen and to remove waste products from the growing tumor. Without a tumor blood supply, incipient neoplasms cannot grow beyond 2 mm 3 .1-3 Termed as the “angiogenic switch,” the change from avascular neoplastic growth to vascularized tumor growth is believed to be mediated by a net shift in favor of pro-angiogenic factors over anti-angiogenic factors in the microenvironment. In addition to being a nutrient and oxygen supply, the tumor vasculature is used by metastatic tumor cells as an entry point into systemic circulation. Finally, growth of micrometastases into to full metastatic disease is also dictated by angiogenesis.
A dynamic microcosm of cellular and non-cellular players exists within the growing tumor mass. Non-tumor stromal cells, such as stromal myofibroblasts, perivascular cells, and inflammatory cells, assist in the overall growth and progression of the tumor.4-6 Furthermore, the extracellular matrix (ECM) surrounding these cellular components provides contextual cues for tumor growth and progression. Although the vasculature is technically a stromal component of the tumor mass, it will be considered as a separate entity in the purposes of this review.
Stromal myofibroblasts and the ECM impart substantial, often pleiotropic, influences on tumor angiogenesis. As vessels innervate the tumor mass, a variety of myofibroblast- and ECM-derived signaling cues and proteolytic factors help recruit blood vessels, assist negotiations with the microenvironment, and stabilize the newly formed vessels. In this review, we focus our attention on complex interplay among myofibroblasts, ECM, and the vasculature within tumor microenvironment, highlighting recent discoveries in this area of active investigation.
The Extracellular Matrix Is a Rich Reservoir of Pro- and Anti-angiogenic Cues
The extracellular matrix is a proteinaceous network of macromolecules that provide structural support to its surrounding cells. The ECM can be broadly categorized into the basement membrane, a specialized 50-nm-thick sheet of ECM molecules on which endothelial cell or epithelial cells reside, and the interstitial matrix, a network of ECM molecules in which cells can be found embedded. Although collagen IV is the major component of basement membranes, fibrillar collagens, such as collage I, II, and III, can be mostly found in interstitial matrices. 7 Along with collagen, other ECM molecules, including laminins, heparan sulfate proteoglycans (HSPGs), and fibronectins, come together to form a mesh-like network. Matricellular molecules, such as tenascins, entactin, thrombospondins, and SPARC (secreted protein, acidic and rich in cysteine), can be found embedded within the ECM.
During tumor development and angiogenesis, the ECM is far more than structural scaffolding. It provides contextual cues to endothelial cells through integrin signaling, affecting processes such as proliferation, differentiation, migration, and survival. Collectively, integrins recognize a variety of ECM and matricellular protein molecules, including collagens, laminins, fibronectins, thrombospondins, and tenascins. Although integrins themselves do not have enzymatic capabilities, integrin activation relays to downstream signaling pathways, such as focal adhesion kinase (FAK), the Src family of kinases, and integrin-linked kinase (ILK). 8 With integrin binding sites removed or cryptic ones exposed by the action of matrix remodeling proteins, integrin signaling is a dynamic sensor to a continuously remodeling microenvironment. 9
Endothelial cells express a limited number of integrins, including α1β1, α2β1 αvβ3, αvβ5, αvβ8, and α5β1. Genetic ablation of integrin subunits αv, β1, β3, β5, or β8 resulted in vascular defects with lethal consequences.10-14 Expression of select integrins coincides with various steps of angiogenesis, suggesting specific functions at these particular steps. 15 Furthermore, although it is well known that integrin signaling controls various processes in endothelial cells, it can also affect tumor angiogenesis by acting on mural cells. 16
The ECM also serves as a rich reservoir for pro-angiogenic and anti-angiogenic factors. Regulating the bioavailability of pro-angiogenic and anti- angiogenic factors is another way the ECM participates in tumor angiogenesis. Vascular endothelial growth factor (VEGF) and basic fibroblasts growth factor (bFGF) are often sequestered by HSPGs in the ECM, limiting their signaling capacity to the local vicinity of their source. Activity from transforming growth factor β (TGFβ) and insulin-like growth factors (IGFs) can be fine-tuned by latent TGFβ binding proteins (LTBP) and IGF binding proteins (IGFBPs), respectively. Both LTBPs and IGFBPs possess domains that bind ECM proteins. Proteolytic activity from matrix remodeling proteins can liberate these angiogenic growth factors. 17
Many matricellular proteins in the ECM also exhibit pro-angiogenic or anti- angiogenic activity through a wide range of mechanisms. SPARC can inhibit tumor angiogenesis by directly binding to angiogenic factors or by altering ECM assembly. 18 Although thrombospondins-1 and -2 can bind to VEGF as well, they can also act directly on endothelial cells to inhibit the angiogenic process via interactions with CD36, CD47, and integrins. 19 In contrast, tenascin-C (tnC) promotes angiogenesis. TnC expression is associated with vascular sprouts in astrocytomas, and loss of TnC resulted in less angiogenic tumors.20,21 Osteopontin can promote an autocrine VEGF signaling loop in endothelial cells.22,23
ECM proteins themselves harbor cryptic anti-angiogenic domains. The non- collagenous (NC1) domains of collagen molecules have potent anti-angiogenic properties. Endostatin, derived from proteolytic cleavage of collagen XVIII, acts by inhibiting endothelial proliferation and migration. 24 It may also act as a feedback inhibitor of certain MMPs. 25 Arresten, canstatin, and tumstatin are derived from NC1 domains of collagen IV α1, α2, and α3 chains, respectively. Although the mode of angiogenic inhibition in arresten and tumstatin is mediated by integrin interactions and blocking VEGF or bFGF-stimulated ERK and Akt signaling, canstatin, through interactions with integrin αvβ3 and αvβ5 and the FasL receptor, potentiates endothelial apoptosis.26,27 Proteolysis of other ECM molecules such as perlecan, fibronectin, and fibulins can produce fragments with anti-angiogenic properties. 28
Several classes of proteases participate in ECM remodeling. The most prominent class is matrix metalloproteinases (MMPs). The angiogenic contribution of MMPs is best illustrated in xenograft studies using MMP-deficient hosts. Here, tumors grew at a slower pace with impairments in angiogenesis.29-32 In particular, MMPs-2, -3, -7, -9, and -16 are believed to play key roles in regulating tumor angiogenesis. 33 Regulation of VEGF bioavailability is mostly attributed to the action of MMP-9 and MMP-7.34,35
MMP activity can be fine-tuned in the tumor microenvironment by endogenously produced tissue inhibitors of metalloproteinases (TIMPs). In mammals, there are 4 TIMPs (TIMPs 1-4). Because MMPs are largely a pro-angiogenic force, TIMPs are, therefore, largely anti-angiogenic. 36 Aside from directly inhibiting MMP activity, TIMPs can have non–MMP-dependent effects on angiogenesis. TIMP-2 can inhibit angiogenesis by inhibiting FGF2-dependent endothelial proliferation via interactions with a3b1 integrin.37,38 TIMP-3 has been found to disrupt the VEGF binding to VEGFR-2. 39
ADAMs (a disintegrin and metalloproteinases) and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) are additional classes of metalloproteinases whose role in tumor angiogenesis is still emerging. In addition to having proteolytic activity for various ECM molecules, ADAMs and ADAMTSs possess a variety of signaling and binding domains, allowing for an expanded repertoire of functional capabilities. These include disintegrin domains, EGF-like domains, and, more specifically in ADAMTSs, thrombospondin type I sequence repeat (TSR) domains. 40 In general, although ADAMs are membrane-bound, ADAMTS are associated with the ECM through its TSR domain. Some ADAM/ADAMTS members lack proteolytic activity.
A pro-angiogenic role of ADAMs has been implicated through the use of an ADAM-specific inhibitor. 41 A splice isoform of ADAM-9, known as ADAM-9-S, can be secreted and can mediate stromal-tumor interactions and degrade laminins at the invasive front of the tumor. 42 More recently, loss of ADAM-17 cells has been demonstrated to affect cell proliferation and vessel formation in vitro. 43 Furthermore, its proteolytic activity is key to tumor necrosis factor α (TNFα) and TGFβ shedding, which may have secondary effects on angiogenesis. 44 ADAM-10 acts as a sheddase for Delta, a key ligand in the Notch-Delta pathway, and may affect tumor angiogenesis in this manner. 45
So far, only 6 ADAMTSs (ADAMTS-1, -2, -8, -9, -12, -15) are known to affect angiogenesis, and all of them appear to exert anti-angiogenic effects.46-49 This is attributed, in part, to their TSP domains, which may directly interact with endothelial cells or sequester the 165-kDa isoform of VEGF. Caution must be taken to not overgeneralize the role of ADAMs and ADAMTSs in tumor angiogenesis, as the potential role of many family members remains unknown.
Tumor angiogenesis is also regulated by the ECM architecture. ECM stiffness, density, and patterning have been implicated in modulating endothelial cell survival, sprouting, and migration.50-53 Although the pro-angiogenic properties of syndecan-1 has been attributed to integrin-based signaling to endothelial cells, it may also contribute in part by reorganizing the structure of fibrillar collagen to promote directional endothelial cell migration.54,55 Needless to say, ECM architecture is not a stagnant feature of the microenvironment, as active remodeling of such ECM parameters by endothelial cells has been captured via in vivo imaging methods. 56
Stromal Myofibroblasts Control Tumor Angiogenesis through Direct and Indirect Interactions
As one of the first cell types recruited to an incipient tumor, stromal myofibroblasts produce a vast array of secreted factors that modulate both tumor and endothelial behavior. Characterized by their spindle-shaped morphological characteristics and expression of smooth muscle actin (αSMA), stromal myofibroblasts are a principal source for growth factors, chemokines, ECM molecules, and matrix-remodeling proteins within the tumor microenvironment.
Not surprisingly, stromal myofibroblasts are often found at the leading edge of the tumor, a place where tumor–host interactions, such as angiogenesis and local ECM remodeling, are most robust. 57 Several studies show increased angiogenesis in xenograft models where tumor cells were co-inoculated with stromal myofibroblasts.58,59 The importance of stromal myofibroblast in tumor angiogenesis is further highlighted in a study demonstrating that recruited myofibroblasts act as a secondary source of VEGF and compensate for the loss of VEGF in tumor cells. 60
Stromal myofibroblasts participate in tumor angiogenesis through a multi-prong approach (Fig. 1). First, they provide a repertoire of secreted pro-angiogenic growth factors, including VEGF, bFGF, TGFβ, platelet-derived growth factors (PDGFs), hepatocyte growth factor (HGF), connective tissue growth factor (CTGF), and interleukin-8 (IL-8).5,60-63 Combined with other sources of pro-angiogenic growth factors in the tumor, myofibroblast-derived pro-angiogenic factors can tip the angiogenic balance in favor of tumor angiogenesis.
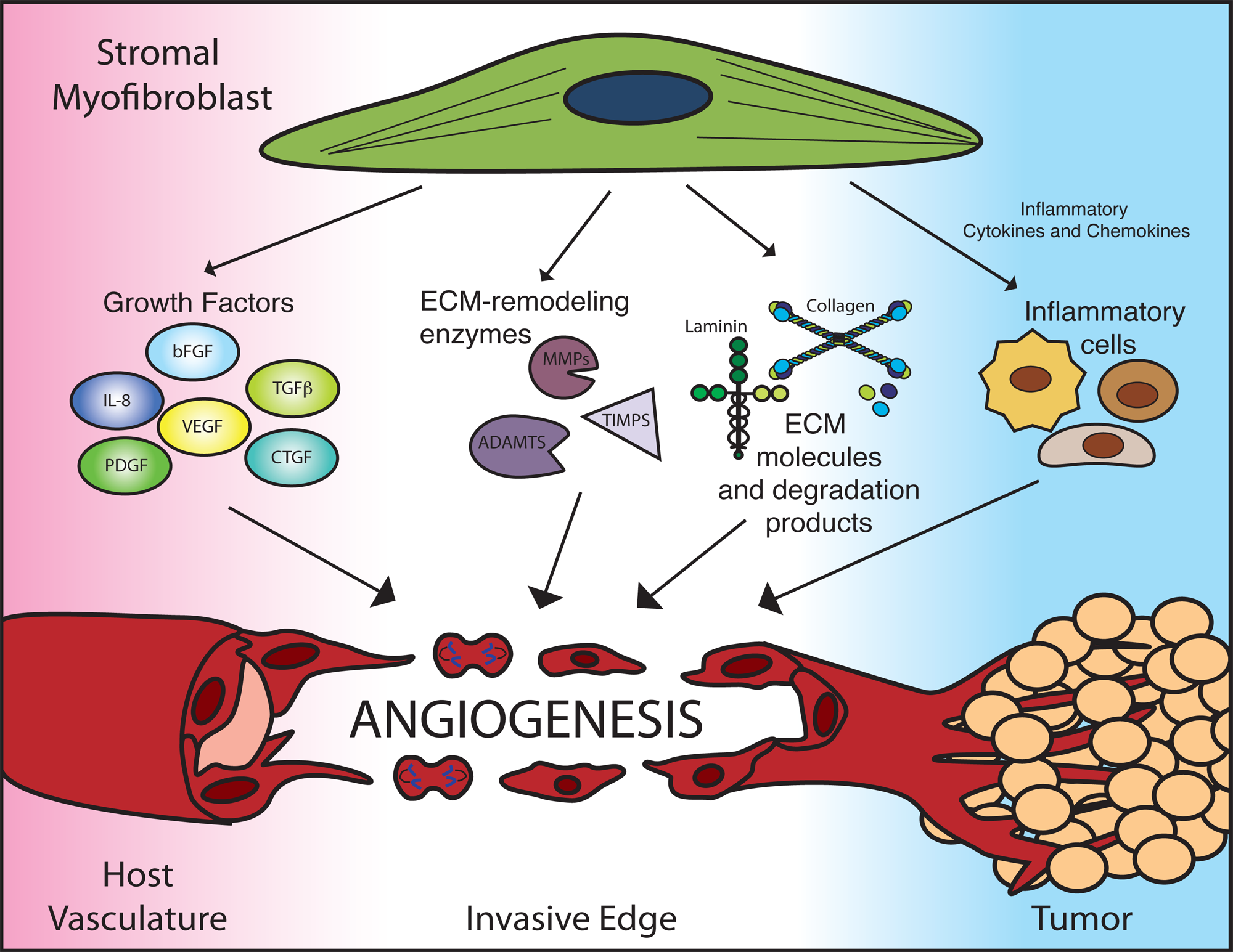
Stromal myofibroblasts modulate angiogenesis with a multiprong approach. Stromal myofibroblasts are a prominent source of angiogenic growth factors, extracellular matrix (ECM) remodeling factors, and ECM components. Additionally, myofibroblast-derived inflammatory cytokines and chemokines recruit infiltrating immune cells, such as macrophages, neutrophils, and T-cells, which can have secondary effects on angiogenesis. ADAMTS, a disintegrin and metalloproteinases with thrombospondin motifs; bFGF, basic fibroblast growth factor; CTGF, connective tissue growth factor; IL-8, interleukin-8; MMPs, matrix metalloproteinases; PDGF, platelet-derived growth factor; TGFb, transforming growth factor b; TIMPs, tissue inhibitor of MMP; VEGF, vascular endothelial growth factor.
Stromal myofibroblasts are a key source of matrix remodeling proteins within the tumor microenvironment, including MMP-1, MMP-2, MMP-3, MMP-7, MMP-11, and MT-MMP1.64-67 Induction of MMPs in stromal myofibroblasts by tumor-derived factor EMMPRIN further stimulates tumor angiogenesis.68,69 In colorectal cancer, ADAMTS-12 expression is selectively robust in myofibroblasts. 70 Furthermore, in the liver, ADAM-9, ADAM-12, ADAM-28, ADAMTS-1, and ADAMTS-2 are up-regulated following activation of hepatic stellate cells, which are stromal myofibroblasts of the liver, either in hepatocellular carcinoma or cirrhotic livers.30,71
In addition to recruiting existing endothelial cells to proliferate, migrate, and form blood vessels, stromal myofibroblasts may elicit endothelial progenitor cells to build blood vessels de novo, a process called vasculogenesis. Secretion of stromal-derived-factor 1 by stromal myofibroblasts (SDF1/CXCL12) stimulates recruitment of Sca1+CD31+ EPCs to the primary tumor. 72
Modulation of the inflammatory response within the tumor microenvironment by stromal myofibroblasts is an additional mechanism to amplify its angiogenic agenda. Stromal myofibroblasts express an array of pro-inflammatory cytokines and chemokines, a process that recruits immune cells to the local microenvironment.72-76 Following recruitment, the secretory milieu of macrophages, neutrophils, and mast cells includes pro-angiogenic factors, such as VEGF, or ECM remodeling proteins, such as MMP-9 and MMP-13. A more detailed overview on the influence of inflammatory cells on tumor angiogenesis can be found in Noonan et al 77 and Zumsteg and Christofori. 78
The genetic landscape of stromal myofibroblasts has gained considerable interest in recent years. Mutations in stromal cells have been implicated in tumor initiation.79,80 Stromal-specific mutations of key tumor-suppressive genes, such as TP53 and PTEN, have been identified in patient samples.81,82 Loss of stromal p53 has been identified critical step in tumor progression in a murine model of prostate cancer. 83 In addition having a direct effect on tumor cells, p53 in stromal myofibroblasts can modulate tumor angiogenesis as well. p53 regulates the expression of TSP-1 and SDF-1, two potent regulators of angiogenesis, in stromal fibroblasts.84,85 As mentioned already, stromal myofibroblasts are a prominent source of VEGF and bFGF, two angiogenic factors regulated by p53.86,87 Dissecting the functional consequences of specific mutations in stromal myofibroblasts will be an exciting area to watch in cancer biology.
Fibrosis and the Desmoplastic Reaction in Tumor Angiogenesis
In many solid tumors, dense fibrotic tissue, rich in myofibroblasts and ECM molecules, often encapsulates proliferating tumor mass. Termed the stromal or desmoplastic response, it is a prominent feature in many cancers, including those of the breast and pancreas. Given their well-characterized role in fibrosis, stromal myofibroblasts are believed to be the predominant orchestrators of the desmoplastic response. 88
The underlying cause of the desmoplastic response in cancer remains largely speculative. Some hypothesize that it is a host defense mechanism against incipient neoplasms, where the fibrotic tissue essentially isolates the neoplastic cells from the rest of the organ. 87 In contrast, because of similarities between wound healing and the stromal response, others have suggested that the tumor cells hijack the wound-healing mechanism to support their own growth.89,90 In concordance with this notion, exposing fibroblasts to tumor cells induces changes in fibroblast gene expression that promote invasion and angiogenesis.91,92
Whether the desmoplastic response promotes or inhibits tumor angiogenesis is up for debate. On one hand, the desmoplastic response coincides with the invasive front of the tumor. An area of active angiogenesis, the invasive front is rich in angiogenic growth factors and ECM remodeling proteins. Furthermore, increased angiogenesis is a feature of fibrosis in certain organs.93,94 On the other hand, excessive deposition of ECM creates a physical barrier that impedes angiogenesis. 95
Studies targeting the desmoplastic process have suggested that an overtly fibrotic microenvironment is counterproductive to angiogenesis. In pancreatic cancer, tumor cell–derived sonic hedgehog (Shh) was found to stimulate myofibroblast activation and desmoplasia formation in pancreatic cancer.96-98 Administration of an inhibitor of sonic hedgehog signaling led to reductions in myofibroblast proliferation, type I collagen, and desmoplastic formation. These changes were correlated with increased angiogenesis and enhanced perfusion throughout the tumor. 99 In another murine model, pharmacological inhibition or genetic deletion of fibroblast activation protein (FAP), a serine protease expressed on fibroblasts and pericytes, has resulted in tumors with increased collagen deposition and decreased angiogenesis. 100
Endothelial Cells in the Tumor Microenvironment Can Affect Neighboring Stromal Components
Stromal myofibroblasts and the ECM are important agents in modulating tumor angiogenesis. They participate in every step of angiogenesis and can be responsible for mediating resistance to chemotherapeutic, anti-angiogenic, and other targeted therapies. However, a clearer understanding of how tumor cells, endothelial cells, and stromal cells co-exist and collaborate is needed.
Although this review has focused on the unidirectional communication from stromal myofibroblast/ECM to endothelium in tumor angiogenesis, one must not forget that endothelial cells may reciprocally affect myofibroblasts and the ECM as well. The most parsimonious manner by which endothelial cells can interact with its stromal partners is through ECM remodeling. During tumor angiogenesis, endothelial cells also produce ECM remodeling proteins to assist in their own navigation through the ECM. Successful establishment of tumor vasculature will reoxygenate hypoxic areas, altering the expression profiles of cells within that area, including that of stromal myofibroblasts. In recent years, it has been demonstrated that endothelial cells can act as niche cells to brain tumor stem cells. 101 Thus, it is conceivable that paracrine signals from vascular cells can alter other cells in the tumor stroma as well. Furthermore, recruitment of bone marrow–derived myofibroblast progenitors, termed fibrocytes, may rely on coordinated presentation of adhesion molecules on the endothelium, perhaps using mechanisms similar to that of leukocyte extravasation. Additionally, endothelial cells themselves can serve as a source of stromal myofibroblasts in the tumor microenvironment by undergoing endothelial-to-mesenchymal transition. 102 Indeed, the influence of the endothelial cells on its surrounding stromal neighbors remains an underexplored area.
Footnotes
Acknowledgements
The authors thank Vesselina Cooke for her critical reading of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by NIH grants CA 125550, CA-155370, CA151925, DK 055001, DK 55001 and the Champalimaud Metastasis Programme.