
Abstract
c-myc promoter binding protein (MBP-1) is a multifunctional protein known to regulate expression of targets involved in the malignant phenotype. We have previously demonstrated that exogenous expression of MBP-1 inhibits prostate tumor growth, although the mechanism of growth inhibition is not well understood. We hypothesized that MBP-1 may modulate microRNA (miRNA) expression for regulation of prostate cancer cell growth. In this study, we demonstrated that exogenous MBP-1 upregulates miR-29b by 5- to 9-fold in prostate cancer cells as measured by real-time quantitative reverse transcription polymerase chain reaction. Subsequent studies indicated that exogenous expression of miR-29b inhibited Mcl-1, COL1A1, and COL4A1. Furthermore, a novel target with potential implications for invasion and metastasis, matrix metallopeptidase-2 (MMP-2), was identified and confirmed to be a miR-29b target in prostate cancer cells. Together, these results demonstrated that exogenous expression of miR-29b regulates prostate cancer cell growth by modulating antiapoptotic and prometastatic matrix molecules, implicating the therapeutic potential of miR-29b for prostate cancer inhibition.
Introduction
Prostate cancer is the most commonly diagnosed cancer in men and one of the leading causes of cancer death in the United States. 1 Although prostate cancer is frequently curable in its early stage by surgical or radiation ablation, many patients present with locally advanced or metastatic disease for which there are currently no curative treatment options. 2,3 Docetaxel-based chemotherapy has recently been introduced for the treatment of metastatic prostate cancer, following demonstration of its survival benefit in advanced hormone-resistant prostate cancer patients. 4,5 However, additional effective therapies with more favorable adverse effect profiles that can cure localized tumors and prevent their metastasis are urgently needed.
Tumor suppressors regulate diverse pathways to block tumor growth. We have shown previously that intratumor injection of c-myc promoter binding protein (MBP-1) inhibits prostate tumor growth in xenograft nude mice and induces cell death in a number of cancer cells without affecting the normal cell growth. 6-8 MBP-1 was originally described to bind to and repress c-myc promoter function, 9 but subsequent investigation has demonstrated that the tumor suppressor function of MBP-1 is not solely dependent on c-myc repression. 7,10,11 However, the mechanism of MBP-1–mediated inhibition of prostate cancer cell growth is poorly understood. We hypothesized that MBP-1 could exert its antitumor action by differentially regulating expression of microRNA (miRNA). miRNA are transcribed genes processed to single-stranded regulatory RNA of ~22 nucleotides. 12 Mature miRNA repress protein expression primarily through base pairing of a seed region with the 3′ untranslated region (UTR) of the target mRNA leading to inhibition of translation and/or mRNA degradation. An individual miRNA is capable of regulating dozens of distinct mRNAs, and together the >650 human miRNA are believed to modulate more than one-third of the mRNA species encoded in the genome. 13 Some miRNAs play a role in growth control or apoptosis, providing a mechanistic underpinning for the relation between miRNA and cancer. 14-16 Moreover, miRNA involved in specific networks, such as the apoptotic, proliferation, or receptor-driven pathways, could likely influence the response to targeted therapies or to chemotherapy. A differential expression of a subset of miRNA between normal tissue and cancer and between cancers has been noted. 14,15 We identified a number of altered miRNA expression upon enforced MBP-1 expression and chose to examine the function of miR-29b induction in prostate cancer cells, which could be a downstream mediator of tumor suppression by MBP-1, because miR-29b plays a role in regulating antiapoptotic and prometastatic proteins.
In this report, we have observed that MBP-1 upregulates miR-29b in prostate cancer cells, which in turn inhibits Mcl-1, matrix metallopeptidase-2 (MMP-2), and collagen expression. These results also add to the growing list of miRNAs that regulate tumor growth and provide a potential mechanism for how MBP-1 suppresses tumor growth by altering the expression of miRNA.
Results
MBP-1 upregulates miR-29b
MBP-1 has already been shown to regulate the expression of multiple genes involved in cell growth metastasis. 7,10 Preliminary microarray analysis comparing the expression profile of miRNA between prostate cancer PC3 cells transduced with AdMBP-1 or control virus (dl312) showed an alteration of the expression of 7 miRNA (R. Ray and R. Steele, manuscript in preparation). We chose to focus our investigation on miR-29b because it was reported to be involved with the regulation of cell proliferation, apoptosis, and migration. Quantitative reverse transcription polymerase chain reaction (RT-PCR) demonstrated miR-29b was significantly upregulated (6- to 8-fold) by enforced MBP-1 expression in PC3 and DU145 prostate cancer cell lines (Fig. 1).
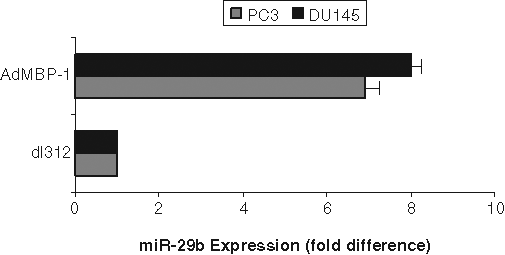
c-myc promoter binding protein (MBP-1) upregulates miR-29b in prostate cancer cells. PC3 and DU145 cells were transduced with control dl312 or AdMBP-1 for 48 hours. Total RNA was isolated, and expression of miR-29b was measured using quantitative reverse transcription polymerase chain reaction with a hydrolysis probe and standardized to small nuclear RNA U6, which was used as the endogenous control. The results are presented from 3 independent experiments, mean ± standard error. P < 0.001 by 1-way analysis of variance.
miR-29b inhibits Mcl-1 in PC3 cells
Mcl-1 is an antiapoptotic Bcl-2 family member protein that is highly regulated in normal cells, and when dysregulated, it contributes to cancer. Enhanced Mcl-1 expression has been observed in multiple human cancers, often in association with poor prognosis, disease recurrence, or drug resistance. 17 miR-29b was shown to directly downregulate translation of the Mcl-1 protein in cholangiocarcinoma cell lines. 17 We tested whether ectopic expression of miR-29b could alter Mcl-1 protein expression in prostate cancer cells. A significant reduction in Mcl-1 protein was observed in the PC3 cell line upon introduction of miR-29b as compared with control miRNA exposed PC3 cells (Fig. 2A). However, we could not detect Mcl-1 expression in DU145 cells. Conversely, anti–miR-29b enhanced Mcl-1 expression in PC3 cells as compared with control anti-miR–treated cells (Fig. 2B). Densitometric scanning of protein bands suggested an ~60% reduction in Mcl-1 protein in the PC3 cells expressing miR-29b, whereas anti–miR-29b–expressing cells display enhanced Mcl-1 expression (~75%). The blots were probed with actin for similar protein load. Densitometric scanning results are presented after normalization of protein load. Furthermore, the signals from Figure 2A cannot be compared with Figure 2B because they were from 2 different gels and exposed for different length of time.
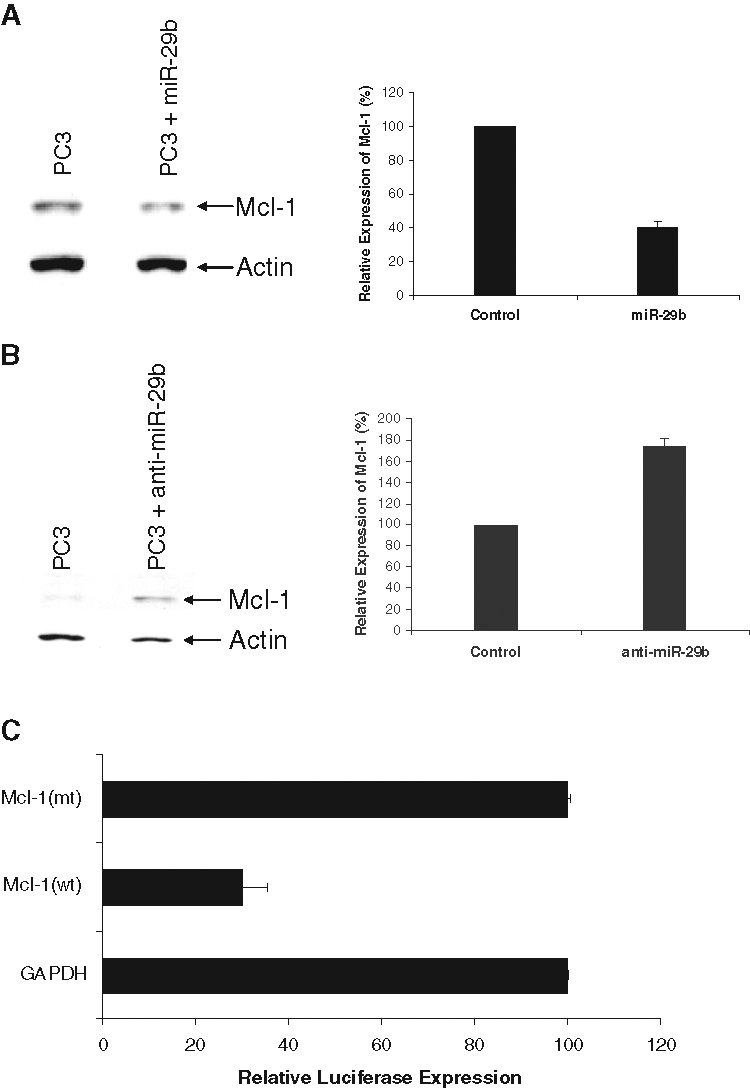
miR-29b modulates Mcl-1 expression. (
To further determine if miR-29b inhibits Mcl-1 3′UTR, we performed the luciferase assay by cotransfecting Mcl-1 (wt) or Mcl-1 (mt) with miR-29b precursor. In parallel, we used the GAPDH 3′UTR, which is not a miR-29b target, downstream of luciferase as a control. Our results showed that expression miR-29b inhibits luciferase activity (70%) of Mcl-1 (wt) but not the mutant form (Fig. 2C). Together, these results suggest that miR-29b may have a cell growth inhibitory effect by altering antiapoptotic protein expression in prostate cancer cells.
Collagens are directly targeted by miR-29b in prostate cancer cells
Recently, the miR-29 family was shown to inhibit collagen mRNA expression in nasopharyngeal carcinomas and osteoblast differentiation. 18,19 To determine if miR-29b regulated collagen expression in prostate cancer cell lines, we used a firefly luciferase reporter containing the relevant miR-29b binding sites (COL1A1, COL3A1, and COL4A1) in the luciferase 3′UTR. In parallel, we used the GAPDH 3′UTR, which is not a miR-29b target, downstream of luciferase as a control. Prostate cancer cells were transfected with these constructs with or without miR-29b mimic transfection. The 3′UTRs of all of these 3 candidate target genes (collagen 1A1, 3A1, and 4A1) elicited significantly decreased luciferase activities in miR-29b mimic transfected cells (Fig. 3). As expected, we did not observe an inhibition of GAPDH 3′UTR. Mutant 3′UTRs of all of these 3 candidate target genes (nucleotide substitutions disrupting the miR-29b binding sites) was also used. miR-29b did not alter the luciferase activity of these mutants. Similarly, control miRNA did not alter the luciferase activity of COL1A1, COL3A1, and COL4A1.
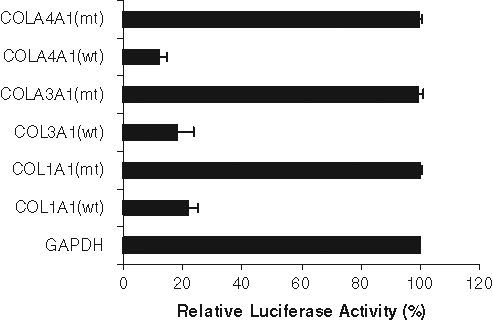
miR-29b inhibits expression of luciferase with 3′UTRs derived from its target genes. 3′UTR of target genes containing miR-29b–binding sites were cotransfected with miR-29b mimic into prostate cancer cells. Luciferase activity was measured after 48 hours of transfection. Luciferase activity from GADPH 3′UTR transfected cells was arbitrarily set as 100%. The results are presented as mean ± standard error from 3 independent experiments. P < 0.001 by 1-way analysis of variance.
miR-29b inhibits collagen mRNA expression in prostate cancer cells
To test whether miR-29b indeed regulates the levels of the candidate target mRNAs, we transfected miR-29b mimic into PC3 cells. Cells were also transfected in parallel with a control mimic miRNA. In PC3 cells, 2 potential miR-29b target mRNAs were reduced significantly by miR-29b mimic transfection: COL1A1 and COL4A1 (Fig. 4). Collagen 3A1 mRNA expression was undetectable in PC3 cells, so its inhibition could not be determined. The reason for lack of collagen 3A1 mRNA in PC3 cells is not clear at present. Specifically, COL1A1 mRNA levels were 35% of control levels, whereas COL4A1 mRNA levels were reduced to 25%. Similar results were obtained when PC3 cells were transduced with AdMBP-1 (data not shown).
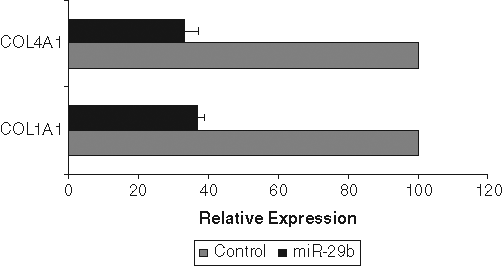
miR-29b downregulates collagen mRNA expression. PC3 cells transfected with control miRNA and miR-29b precursor for 48 hours. Total RNA was isolated and COL1A1 and COL4A1 mRNA was quantified using reverse transcription quantitative polymerase chain reaction. GAPDH was used as an internal control and normalized for relative mRNA expression. mRNA from control miRNA transfected cells was arbitrarily set as 100%. The result presented was the mean from 3 independent experiments. P < 0.001 by 1-way analysis of variance.
miR-29b inhibits MMP-2 expression
MMP-2, a zymogen requiring proteolytic activation for catalytic activity, has been implicated broadly in invasion and metastasis of many cancer model systems, including prostate cancer. 20-22 MMP-2 has been shown to be activated by collagen type 1, 23 and once activated, it degrades type 4 collagen in the basement membrane. Using the Targetscan computer prediction method, a putative miR-29b target site in MMP-2 3′UTR was noted (Fig. 5A). To examine whether miR-29b inhibits MMP-2 expression, we performed Western blot analysis in control or miR-29b mimic transfected PC3 cells. Our results demonstrated a significant downregulation of pro-MMP-2 in miR-29b–transfected PC3 cells (Fig. 5B). To determine the effect of miR-29b on MMP-2 mRNA level, total RNA was isolated from PC3 cells transiently or stably transfected with the mimic miR-29b or miR-29b expression construct. Control miR was used in parallel. MMP-2 message was quantitated by real-time RT-PCR. We did not observe a significant difference in mRNA level between control and cells expressing miR-29b. Therefore, it appears that miR-29b does not affect MMP-2 mRNA expression. On the other hand, MMP-2 promoter activity was downregulated by MBP-1 in a dose-dependent manner. 10 Thus, these cancer-relevant targets could be repressed directly by MBP-1, miR-29b, or both. Together, this result suggested that miR-29b mediated pro-MMP-2 inhibition may serve as an additional layer of repression of MMP-2 function.
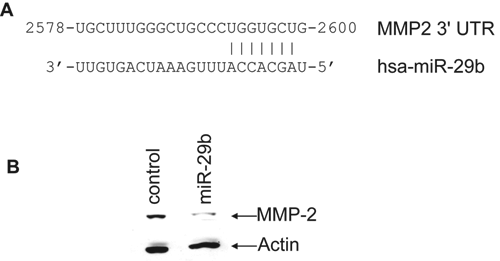
miR-29b targets matrix metallopeptidase-2 (MMP-2) and represses pro–MMP-2 protein expression. (
Discussion
miRNAs play a role in growth control or apoptosis, and a differential expression of a subset of miRNA between normal tissue and cancer and between cancers has been noted. 14-16 We hypothesized that MBP-1, a transcriptional co-repressor that regulates the expression of multiple genes, might directly or indirectly regulate expression of miRNA that modulate the cancer phenotype. Indeed, we have demonstrated that MBP-1 stimulates miR-29b expression in 2 prostate cancer cell lines. miR-29b is of interest because it is involved with regulation of apoptosis and prometastatic molecules with which MBP-1 was already implicated. Preliminary study also suggested that the basal expression level of miR-29b is decreased in 3 of 4 prostate cancer specimens examined as compared with nontumorigenic benign prostatic hyperplasia specimens (R. Ray and G. Deng, unpublished observation). The salient findings of the current study address the molecular mechanisms whereby the MBP-1/miR-29b axis could regulate prostate cancer malignant behaviors. Specifically, in prostate cancer cell lines, the MBP-1–induced miR-29b regulates 1) Mcl-1, 2) collagens, and 3) MMP-2. Each of these targets will be addressed below.
The antiapoptotic protein Mcl-1 is frequently overexpressed in cancers by various mechanisms, including genomic amplification, 24 transcriptional induction, posttranscriptional regulation, 25 and loss of microRNA function. 26 Mcl-1 protein is found at elevated levels in prostate cancer, 27 with more Mcl-1–positive cells in Gleason grade 8 to 10 tumors than lower grade tumors. 28 Furthermore, Mcl-1 induction in prostate cancer cells by the inflammatory cytokine IL-6 provides resistance to apoptosis. 29 Thus, Mcl-1 in prostate cancer, as in other malignancies, is a central mediator of the resistance to apoptosis. In this study, we have observed that MBP-1–induced miR-29b inhibits Mcl-1 protein levels, consistent with previous work. 26
Increased extracellular levels of collagens have been shown to induce increased invasiveness in culture and increased metastasis in animal models. Similarly, increased levels of collagens have been associated with an increased likelihood of clinical metastasis of multiple human solid tumors. 30 Using laser capture isolated tumor cells, Sengupta and others 18 have shown that nasopharyngeal carcinoma cells upregulate collagen mRNAs. Collagen type I contributes to invasiveness and metastasis in pancreatic cancer. 31,32 Furthermore, in prostate cancer, the alpha-1 chain of type IV collagen (COL4A1) is produced by malignant cells and increased with increasing tumor grade, 33 suggesting a role in tumor pathology. Thus, our finding that miR-29b regulates COL1A1, COL3A1, and COL4A1 is consistent with enhanced collagen production upon miR-29b repression, potentially facilitating metastatic activity. Conversely, MBP-1 induction of miR-29b and inhibition of collagen production may present a useful therapeutic goal.
MMP-2 is a key enzyme in the process of extracellular matrix remodeling involved in tumor invasion and metastasis. In hepatocellular carcinomas, increased collagen I mRNA levels were strongly associated with those of MMP-2. 34 Collagen I induces MMP-2 activation in breast cancer cells and hepatocellular carcinoma. 24,34 We demonstrated here that MMP-2 is a predicted target of miR-29b, and we recently showed that MBP-1 expression reduced active MMP-2 in breast cancer cells. 10 Here, we have observed that miR-29b inhibits pro–MMP-2 protein expression in PC3 cells. Although MMP-2 activation is directly inhibited by MBP-1, inhibition by miR-29b could be another mechanism, and a working model is proposed (Fig. 6). This possibility is supported by the observation that expression in MBP-1 in PC3 cells upregulated miR-29b and enforced miR-29b expression downregulated MMP-2. MMP-2 is a protein with significance in prostate cancer, as MMP-2–mediated degradation of type IV collagen in the basement membrane may promote metastatic invasion. Thus, although both the alpha-1 chain of collagen IV and the type IV metalloproteinase MMP-2 are targeted by miR-29b, the end result of low miR-29b may be increased turnover of basement membranes consistent with active remodeling of the extracellular matrix.
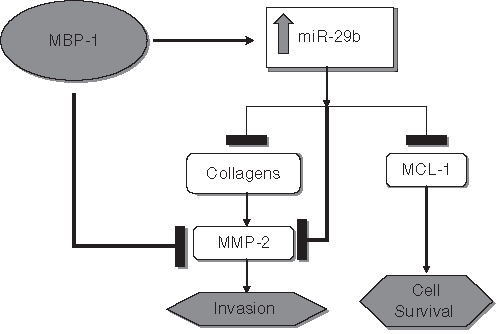
A schematic diagram showing the hypothetical cross-regulation of c-myc promoter binding protein (MBP-1), miR-29b, Mcl-1, collagen, and matrix metallopeptidase-2 (MMP-2) in inhibition of prostate cancer cell growth and invasion. The transcriptional co-repressor MBP-1 inhibits MMP-2 activation while also inducing miR-29b. miR-29b acts to repress Mcl-1, collagens, and pro–MMP-2 protein expression. Type I collagen normally promotes MMP-2 activation; thus, the combined repression of type I collagen and pro–MMP-2, plus MBP-1–dependent inhibition of MMP-2 activation, efficiently prevents its activation. Mcl-1 targeting by miR-29b meanwhile limits cellular survival and thus tumor growth.
The mechanism by which MBP-1 induces miR-29b was not investigated. As miR-29b is repressed by c-myc expression, 35 it is plausible that MBP-1 decreases c-myc in PC3 and DU145 cells, allowing the de-repression of mir-29b. Further experiments will be necessary to confirm this hypothesis. In conclusion, our results demonstrated a mechanistic insight into miRNA function in prostate cancer cells. We have shown that miR-29b targets antiapoptotic and prometastatic molecules in prostate cancer cells for regulation of growth. These results suggest that further preclinical trials are needed to establish whether miR-29b could also be useful alone or in combination with conventional chemotherapeutics or conventional targeted agents for the treatment of prostate cancer.
Materials and Methods
Cell lines and cell culture
PC3 and DU145 cell lines were purchased from American Type Culture Collection and maintained in DMEM containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified CO2 incubator.
Western blot analysis
PC3 and DU145 cells were transduced with a replication deficient adenoviral vector expressing MBP-1 (AdMBP-1; 7) or dl312 control virus, and cell lysates were prepared 48 hours postinfection in 2x SDS-sample buffer. Similarly, cells were transfected with miR-29b mimic or control miRNA (random sequence Pre-miR molecules; Applied Biosystems, Foster City, CA), and cells lysates were prepared 48 hours posttransfection. In addition, anti–miR-29b (Applied Biosystems) was transfected and cell lysates prepared for immunoblot analysis at 48 hours. Mcl-1 or MMP-2 expression was analyzed by Western blot using specific rabbit polyclonal antisera (Santa Cruz Biotechnology, Santa Cruz, CA), followed by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) as described previously. 10 Relative expression level was determined by densitometry, normalized to the expression of actin (mouse monoclonal; Santa Cruz Biotechnology).
Quantitative real-time RT-PCR
miRNA expression was determined by isolating total RNA using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA (200 ng) was reverse transcribed using the TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems) with miRNA-specific primers. To quantify the miRNA levels, the 7500 Real-Time System (Applied Biosystems) was used in conjunction with gene-specific TaqMan assay kits (Applied Biosystems) for miR-29b and U6. U6 was used as an endogenous control to normalize expression. Relative miR-29b expression and standard error were calculated by the supplied 7500 Real-Time System software.
Luciferase assay
Luciferase reporter constructs containing the putative miR-29b binding sites from Mcl-1,
26
Col1A1, Col3A1, and Col4A1
18,19
were as described previously. Constructs were co-transfected into PC-3 or DU145 cells cultured in 12-well plates using Lipofectamine 2000. Briefly, 30 n
Footnotes
Acknowledgements
The authors thank Heather Lavezzi and Biswajoy Pal, the rotation students, for their involvement in the beginning of this work. They thank Dr. Ahlquist, University of Wisconsin, Madison, and Dr. Lian, University of Massachusetts, Worcester, for providing us the 3′ UTR collagen constructs and Dr. Sailen Barik, University of South Alabama, Mobil, for helpful suggestions. They also thank the members of the Ray Laboratory for critical reading of the article.
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the Doisy Research Fund to R.B.R. from Saint Louis University and by a Career Development Grant to J.L.M. (K01-DK79875).