
Abstract
The tumor microenvironment is considered pro-oncogenic for reasons that, among others, involve the stimulation of cancer cell growth. However, little is known regarding how the tumor microenvironment affects the proliferation of stromal cells that coexist with cancer cells in the tumors. In the present study, we show that cancer cells trigger a paracrine response in normal fibroblasts that is not consistently mitogenic but may also become antimitogenic, inhibiting fibroblasts’ proliferation in vitro. In vivo experiments on xenografts of MDA-MB-231 breast and A549 lung cancer cells in SCID mice or primary Notch-induced breast cancers that develop in transgenic mice showed that stromal cells frequently undergo senescence, confirming the presence of stress-inducing conditions within the tumor’s microenvironment. These antimitogenic responses of the stromal cells were less pronounced in fibroblasts that are p53-deficient, suggesting that p53 plays a role in the execution of these mitogenic stress responses. In addition, they provide a biological basis for the selection of certain stromal cell subpopulations within malignant tumors exemplified by the p53-deficient stromal cells. Indeed, reconstitution of A549 human lung tumors in mice with admixed wild-type and p53-deficient stromal fibroblasts indicates that the selection of p53-deficient stromal cells is more intense when the number of cancer cells, and therefore the intensity of the stress response–inducing signals, is higher. These findings possess implications related to the effects of the cancer cells in the stromal cells and provide hints for the mechanism(s) underlining the transition of the normal stroma to cancer-associated stroma.
Introduction
Malignant tumors consist of neoplastic cancer cells and stromal cells. During the development of malignancy, the tumor microenvironment progressively changes and becomes “fixed” in a cancer-associated state that ultimately favors and further promotes tumor growth.1,2 These concomitant and progressive changes between the epithelium and the fibroblastic stroma are orchestrated by the establishment of reciprocally operating communication networks between the cancer and the stromal cells that dictate changes in each other by mechanisms that, as yet, remain poorly understood.1,2
Following the recognition of the stroma as a major regulator of tumorigenesis, much of recent research is focused on the specific changes that occur in the stromal cells, especially the fibroblasts, since these changes are responsible for their commitment to this cancer-associated state. Considerable effort was also made on the characterization of the specific molecular cues that are triggered by the cancer-associated fibroblasts and that stimulate cancer cell growth. These changes involve the neo-expression of specific structural proteins, such as α-smooth muscle actin,3,4 the downregulation of cell cycle and stress response genes, such as p21 and p53, and others.5-7 The elevated production and secretion of various growth factors have also been recognized in stromal fibroblasts, such as stromal cell–derived factor 1,8,9 hepatocyte growth factor, 10 and others, which stimulate the growth of cancer cells. In addition to the specific changes in gene expression, DNA alterations in stromal cells have also been detected and involve tumor suppressors such as p53 and PTEN.11-13 While, however, the latter alterations, when present, have been shown to affect significantly the profile of tumorigenesis as well as the efficiency of chemotherapy,8,14,15 how such lesions are selected within the otherwise genetically stable—in principle—stromal cells, and eventually become detectable in primary human and mouse tumors, remains poorly understood. It is plausible though that this selection of specific molecular lesions in stromal fibroblasts must be associated with their coexistence with the cancer cells since the acquisition of considerable differences in the mutational rates between fibroblasts of a normal versus cancer-associated stroma is not likely to occur. It is also conceivable that this selective pressure in the stromal fibroblasts is associated not only with the clonal expansion of specific lesions that may have randomly occurred in the stroma, such as in the p53 tumor suppressor gene, but is also related to the transition of the normal stromal fibroblasts to a cancer-associated state.
In the present study, we have focused on the effects of the cancer microenvironment on the stromal cells, in the presence or absence of functional p53. Our results suggest that the cancer microenvironment can be antimitotic for the normal stromal cells and also that p53-deficient fibroblasts are consistently less vulnerable to this effect, acquiring a distinct selective advantage. This response is consistent with the induction of a mitotic stress response that is inflicted by paracrine mechanisms by the cancer cells and may also trigger the onset of cellular senescence in the stromal fibroblasts.
Results and Discussion
p53 regulates the mitogenic response of fibroblasts in cancer cell conditioned media and epidermal growth factor (EGF)
Initially, we sought to explore the effects of conditioned media from cancer cells in the rate of cell proliferation of fibroblasts in relation to their p53 status. To that end, mouse embryonic fibroblasts (MEFs) from wild-type or p53-deficient mice were cultured in the presence of conditioned media from A549 lung or MDA-MB-231 breast human cancer cells. Considering that the presumable effects on fibroblasts’ proliferation are mediated by soluble growth factors that are released by the cancer cells, we have collected conditioned media from confluent or semiconfluent (at about 50%) A549 or MDA-MB-231 cells. We have hypothesized that the amount of these soluble factor(s) would have been higher in the confluent as compared to the semiconfluent cells, thus permitting the assessment of potential dose dependency on the proliferation rates we would record. As shown in Figure 1A, media from both A549 and MDA-MB-231 cells were more mitogenic for the MEFs that did not express functional p53, suggesting that the presence of functional p53 suppresses the stimulatory effects of cancer cells’ conditioned media. Interestingly, while media from both confluent and semiconfluent A549 cells were consistently less mitogenic than control media, regardless of the p53 status of the MEFs, the MDA-MB-231 conditioned media collected by both confluent and semiconfluent cells were significantly mitogenic only for p53-deficient fibroblasts. In wild-type fibroblasts, however, the MDA-MB-231 conditioned media only produced a minor, notwithstanding insignificant, stimulatory effect only when collected from semiconfluent but not confluent breast cancer cells. These findings prompted us to hypothesize that specific mitogenic signals are elicited by the cancer cells onto the fibroblasts but may become toxic at certain concentrations. Furthermore, these results are consistent with a role for p53 in the interpretation of this response, according to which functional p53 may sensitize fibroblasts, rendering them more susceptible to the transition of these signals from mitogenic to toxic. In order to test these hypotheses, we have exposed MEFs and lung fibroblasts (LuFi) to increasing concentrations of a growth factor such as EGF that is frequently produced by human cancer cells. As shown in Figure 1B, EGF at increasing amounts caused a dose-dependent stimulation that at concentrations greater than 100 ng/mL was succeeded by dose-dependent suppression of fibroblasts’ proliferation. In all cases, p53 deficiency caused an elevation of the mitogenic response at equal concentrations of EGF. Thus, the mitogenic response that is elicited by a growth factor such as EGF may cause mitogenic stress at high concentrations, inhibiting cell proliferation. In the absence of p53, this antimitogenic response is less pronounced.
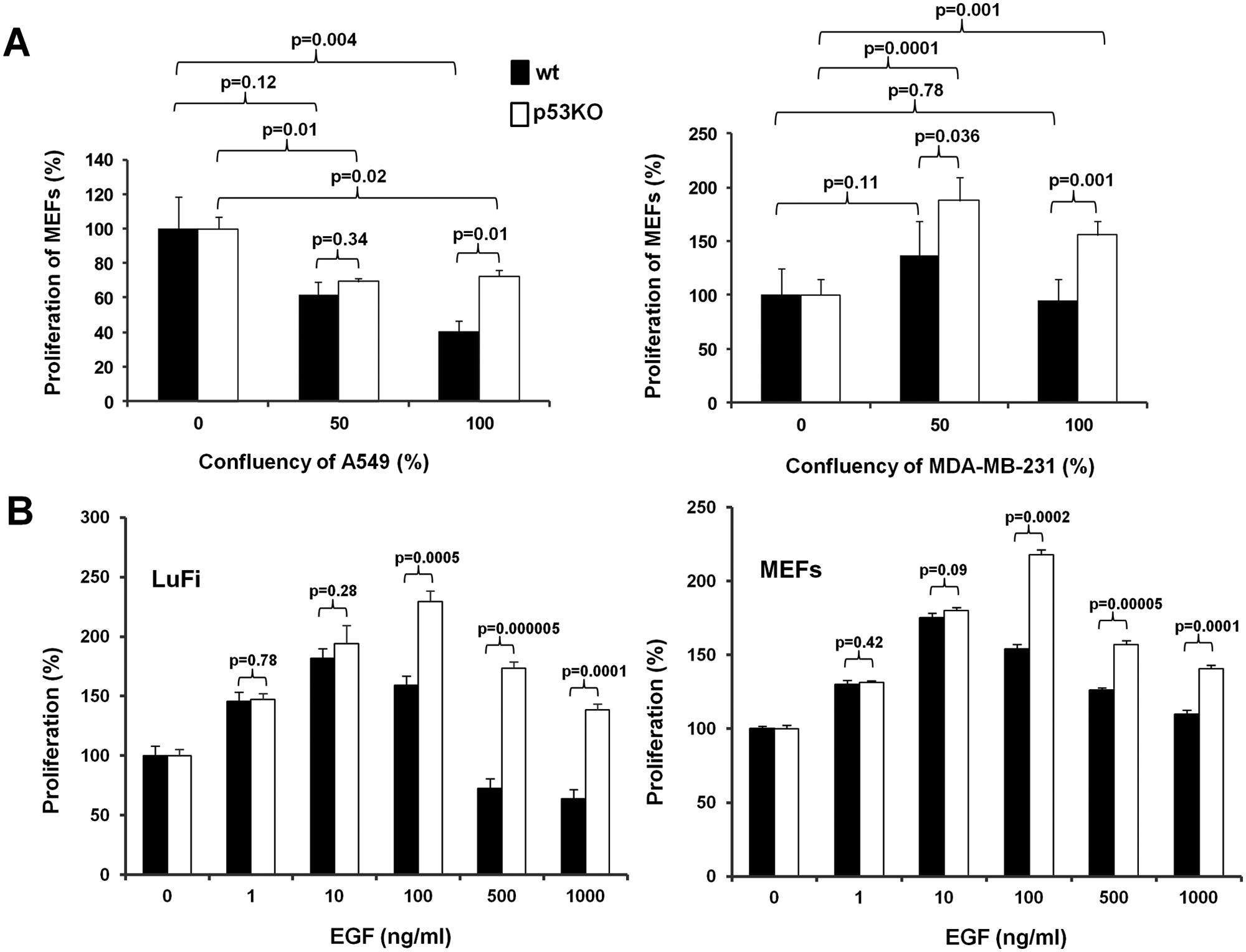
Proliferation of primary mouse fibroblasts in conditioned media from cancer cells and at increasing concentrations of EGF. (
Selection of p53-deficient stromal cells in vivo
That fibroblasts differing in p53 status exhibit differential sensitivity to increasing mitogenic signals elicited by the cancer cells or by a specific growth factor such as EGF in vitro prompted us to explore if this could be interpreted as differential selective pressure, selecting one fibroblast subpopulation versus the other within the tumor microenvironment. Therefore, we have reconstituted human lung tumors in SCID mice with increasing numbers of A549 cells admixed with a stable number of wild-type and p53 knockout (p53KO) cells at a ratio of 1:1 (p53KO:wild-type). Subsequently, after 3 weeks following cell inoculation, DNA was extracted from the xenografts, and mouse wild-type and mutant p53 alleles were amplified by allele-specific PCR. The PCR oligos utilized here were first confirmed to amplify only mouse and not human p53 alleles (Fig. 2). We noted that the signal corresponding to the mutant p53 allele was more prominent in higher A549 cells containing tumors, suggesting that lung cancer cells select for the p53 mutant fibroblasts (Fig. 2). We also noted that in tumors initiated by a low number of A549 cells (106), the wild-type p53 band was more prominent than the p53KO band. This is likely due to the fact that the source of the wild-type p53 alleles is not only from the inoculated fibroblasts but also from the mouse host (Fig. 2). Thus, in the tumors originating from the relatively smaller number of cancer cells, the wild-type alleles predominate.
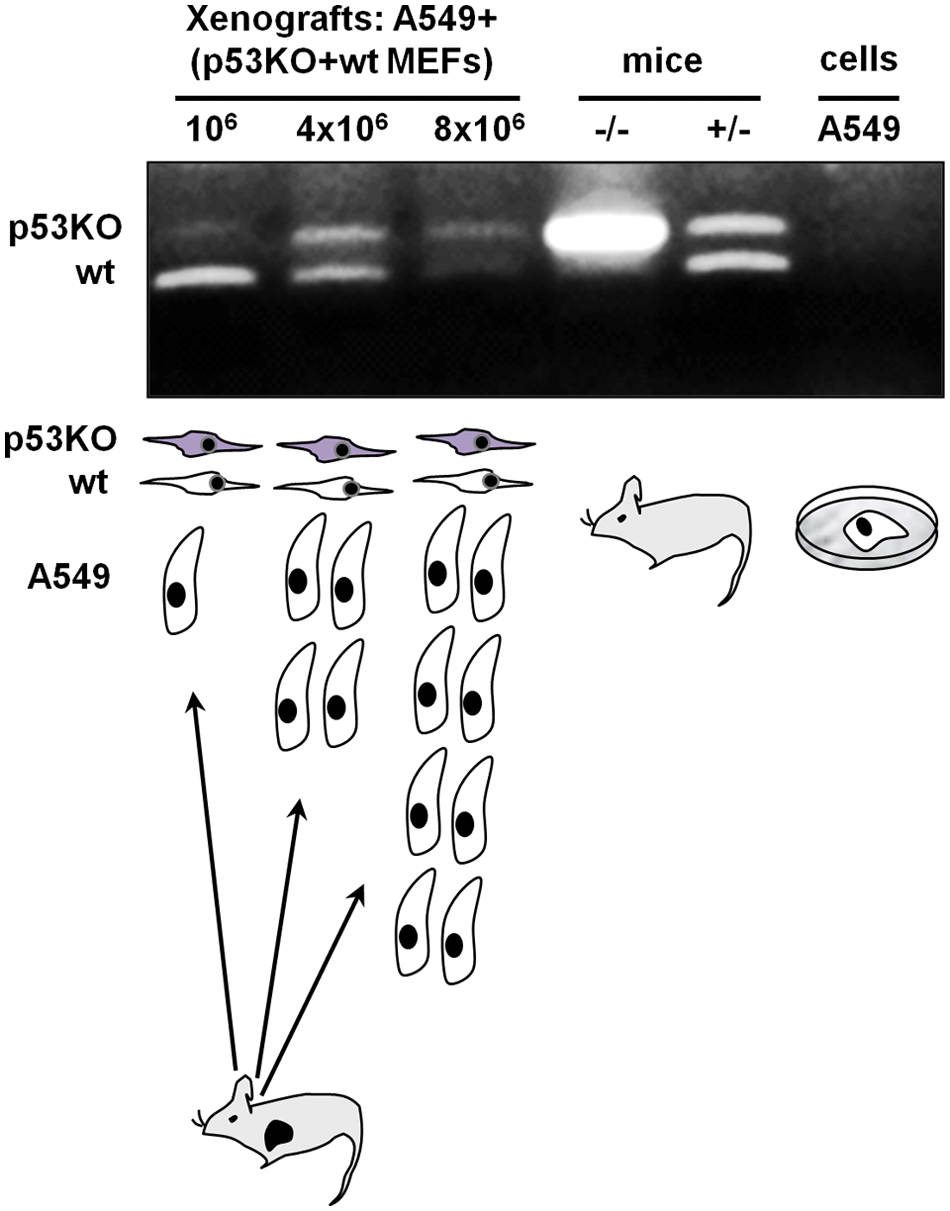
Selection of p53-deficient fibroblasts in vivo at increasing numbers of tumor-initiating A549 lung cancer cells. Allele-specific PCR analysis for wild-type and p53 mutant alleles indicates a higher prevalence of p53KO fibroblasts at higher numbers of A549 cells. The source of the DNA template for the PCR analysis is indicated above the microphotograph of the gel, while the experimental design is shown below the gel microphotograph. When A549 cells cultured in vitro were used as a template, no PCR product was obtained confirming the specificity of the allele-specific PCR for mouse p53.The result of 1 of 3 representative independent experiments is shown.
Induction of senescence in stromal cells in vivo
Stress-inducing stimuli may induce cell cycle arrest and senescence. In order to evaluate if the stress induced by the tumor microenvironment is sufficient to stimulate cellular senescence in vivo in the stromal fibroblasts, xenografts of MDA-MB-231 and A549 cells were assessed for the expression of senescence-associated β-galactosidase (SA-β-gal). Consistently with earlier findings on the induction of senescence in primary ovarian cancers, 16 and the tumor-promoting effects of senescent fibroblasts in vivo, 17 we found that stromal cells and particularly fibroblasts that are located around the tumor cells were occasionally SA-β-gal positive. In Figure 3, the characteristic for SA-β-gal blue staining is shown in the cytoplasm of spindle cells, which represent the above-mentioned fibroblasts. In order to rule out that this induction of senescence in the xenografts represents a methodological artifact of our artificial xenograft-based experimental system, we have also attempted to detect the presence of senescent stromal cells in primary breast cancers that develop in transgenic mice. To that end, we utilized MMTV-hN1IC transgenic mice that during lactation develop benign breast lesions, while after repeated rounds of pregnancy lactation, they develop invasive mammary adenocarcinomas. 18 Consistently with our above-mentioned findings on the xenografted tumors, senescent stromal fibroblasts were detected in primary breast adenocarcinomas that developed in MMTV-hN1IC transgenic animals (Fig. 4). Interestingly, in the tumor-free mammary glands of MMTV-hN1IC mice, SA-β-gal positivity in stromal cells was minimal, implying that it is not the oncogene expression per se but rather the malignant transformation that stimulates senescence in adjacent stromal fibroblasts (Fig. 4).
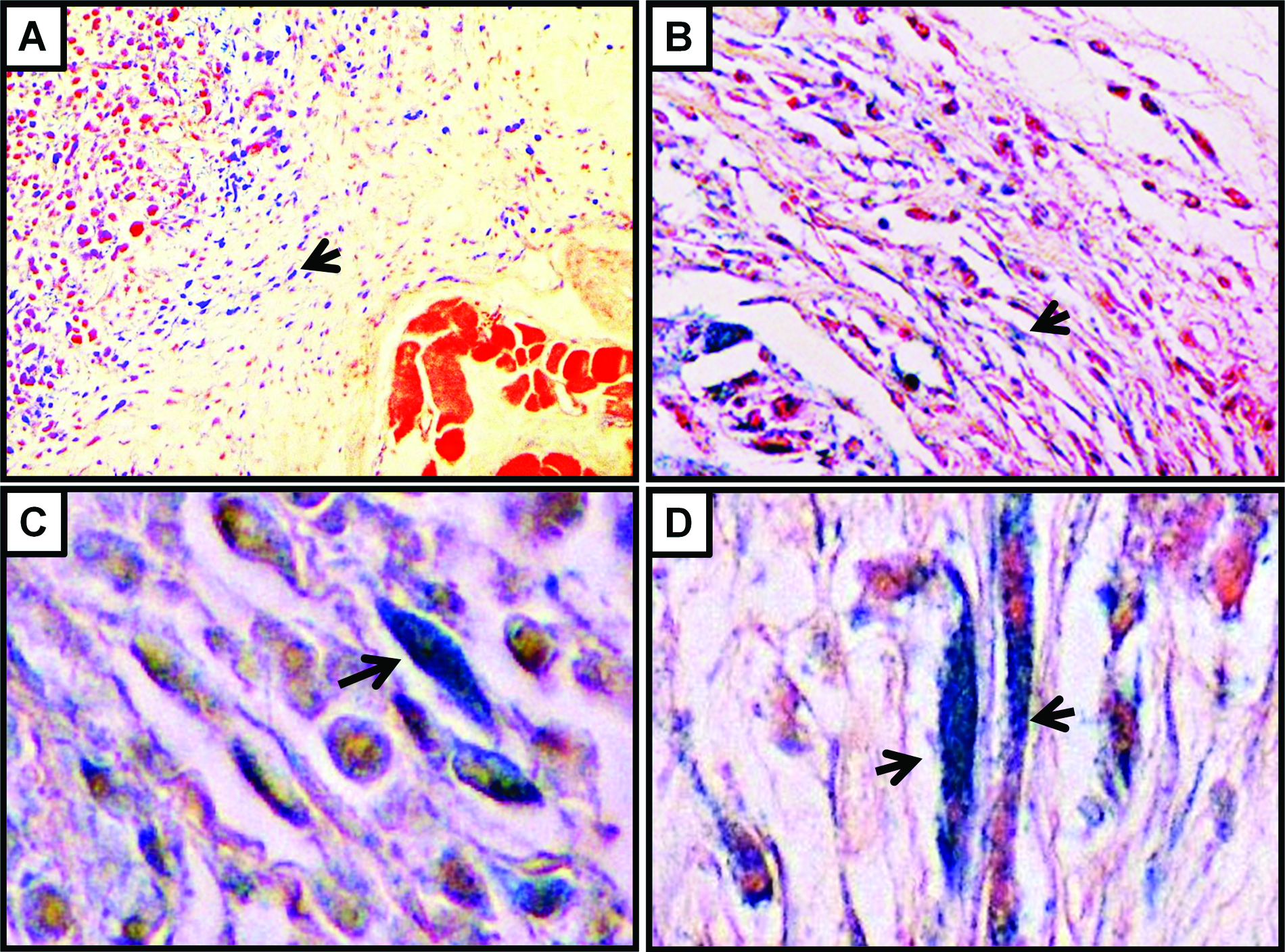
Induction of senescence (SA-β-gal positivity; blue staining indicated by the arrows) in xenografts of MDA-MB-231 (
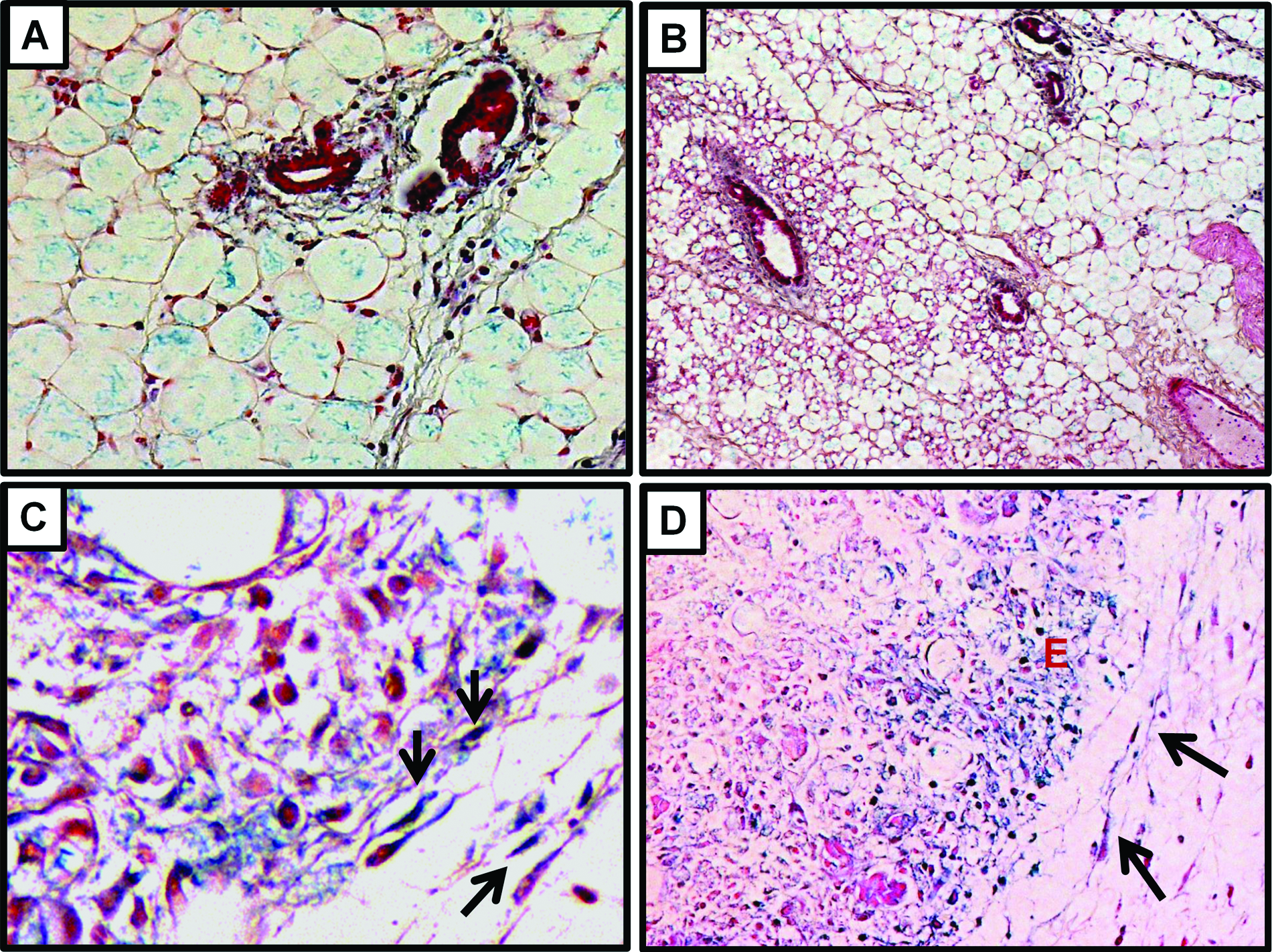
SA-β-gal staining of MMTV-hN1IC mouse mammary tissue from 8-week-old virgin females (
Despite the well-established tumor-promoting role of the stromal fibroblasts in carcinogenesis, little is known on how normal fibroblasts respond when exposed to the cancerous microenvironment of a developing tumor. In the present study, we show that increasing concentrations of a growth factor such as EGF and conditioned media from breast and lung cancer cells elicit mitogenic responses that can be toxic and that are more pronounced in the absence of functional p53. Considering the role of p53 as a nodal point for the interpretation of various and heterogenous stress responses, it is likely that acute mitogenic stimuli trigger mitogenic stress from which p53-deficient cells may escape and acquire a proliferative advantage. This is consistent with the fact that both high concentrations of EGF and A549 conditioned media inhibited the proliferation of normal and, to a lesser extent, p53-deficient fibroblasts. Furthermore, breast cancer cells’ MDA-MB-231 conditioned media were significantly mitogenic in the p53KO fibroblasts only, and this mitogenicity was less pronounced when conditioned media were collected from confluent as compared to semiconfluent cells. Considering that in primary human tumors, the concentration of various growth factors may reach relatively high levels at specific anatomic locations or niches, it is conceivable that this differential sensitivity between the wild-type and the p53-deficient cells may represent a relevant biological mechanism that can explain the selection of the p53 mutant stromal fibroblasts, which have been detected in vivo in primary human tumors. Furthermore, it shows that the strong selective pressure that is applied by the tumor microenvironment on the stromal fibroblasts may select distinct subpopulations of the stromal cells with altered properties. This selection process may in turn provide a bottleneck that can contribute to the transition of the stroma to a cancer-associated state. Indeed, direct evidence for this selective pressure was provided by our observation that higher numbers of cancer cells, and thus the application of stronger selective pressures, offer a stronger proliferative advantage on the p53-deficient fibroblasts as compared to their wild-type counterparts.
It is noted that it is not only mutational inactivation of p53 that may select for specific stromal fibroblasts’ subpopulations. It is conceivable that mutations in other genetic loci as well as epigenetic phenomena may also facilitate the selection of stromal cells that can survive and expand more efficiently within the tumor microenvironment. Consistently with this notion, cancer cell–mediated suppression of p53 and p21 expression has been demonstrated in adjacently located fibroblasts,5-7 while evidence for altered methylation of stromal fibroblasts has also been reported. 19 In favor of the fact that the commitment of fibroblasts to the cancer-associated state is irreversible, persisting prolonged culture, and does not represent a consequence of specific changes in gene expression are earlier findings showing that cultures of fibroblasts from prostate cancers in vitro were not sufficient for the abolishment of their cancer-associated tumor-promoting state. 20
The observation that stromal cells of malignant tumors may develop senescence possesses dual consequences: First, it is related to the previously discussed selection of stromal cells that due to the acquisition of specific molecular lesions, epigenetic alterations, or even stochastically, are more likely to escape these antimitogenic effects and senescence. Second, considering that senescence is specific, and likely other forms of cellular stress as well, triggers a paracrine response that by non–cell-autonomous mechanisms may stimulate cell growth. Thus, cancer cells dictate fibroblasts to enter a state at which the release of mitogenic factors is induced, facilitating their growth. Furthermore, these findings may also have consequences in the regulation of anticancer drug efficacy. Tumor stroma has been found to modulate anticancer therapy as evidenced by the fact that tumors bearing p53 mutant fibroblasts 15 as well as p53-deficient endothelium 21 were more sensitive to conventional cytotoxic therapy. Thus, it is conceivable that the selection of specific molecular lesions present in stromal cells may affect therapeutic efficacy. Finally, even therapy as such will increase the selection of particular lesions in the stroma, especially those that confer resistance to genotoxic stress.
In summary, we have shown that the tumor microenvironment induces strong selective pressure onto the stromal cells, selecting specific subpopulations of stromal fibroblasts. The latter may be also related to the transition of fibroblasts to a cancer-associated state.
Materials and Methods
Cell culture and proliferation assays
A549 human lung epithelial adenocarcinoma cells and MDA-MB-231 human breast adenocarcinoma cells were maintained in DMEM containing 10% FBS and antibiotics/antimycotics. Cell culture reagents were from GIBCO-Invitrogen (Life Technologies, Carlsbad, CA, USA). The MEFs were isolated at E11.5 from female p53 heterozygous mice that have been mated with heterozygous males, by using standard procedures, and were subsequently genotyped to assess the p53 zygosity status as previously described. 14 Primary mouse LuFi were isolated from adult wild-type or p53KO mice using standard procedures. Fibroblasts were maintained in DMEM containing 10% FBS and antibiotics/antimycotics and were used in all assays at early passage.
For conditioned media mitogenic assays, wild-type or p53KO MEFs were plated on 12-well plates at a density of 4 to 8 × 103 cells/well and cultured for 16 hours in serum-free DMEM and then for an additional 48 hours in conditioned media from semiconfluent (about 50%) or confluent A549 or MDA-MB-231 cells. Fibroblasts were counted 48 hours later using the trypan blue exclusion assay.
For the proliferation assay after EGF treatment, wild-type or p53KO MEFs or LuFi were plated on 12-well plates at a density of 104 cells/well, starved in serum-free culture media for 16 hours, and then treated with EGF (Sigma Aldrich, St. Louis, MO, USA) at the indicated concentrations for 6 days. Cell number was assessed using the trypan blue exclusion assay.
Each experiment was performed in triplicate, and average values were calculated by counting cells in at least 3 high power optic fields per experiment. Statistical analysis of the results was performed using the Student t test.
Animal studies
p53 mice (for MEF isolation) 22 and SCID mice (for tumor reconstitution experiments) were originally obtained from Jackson Laboratories (Bar Harbor, ME, USA) and subsequently bred and maintained at the University of Athens. Six- to eight-week-old animals were used for all experiments.
MMTV-hN1IC mice (for development of primary tumors) have been described previously and were subsequently bred and maintained at the University of Athens. 18
For tumor reconstitution experiments, 4 × 106 A549 or MDA-MB-231 cells or increasing numbers of A549 cells (106, 4 × 106, 8 × 106) admixed with 106 wild-type MEFs and 106 p53KO MEFs were resuspended in 0.1 mL of serum-free DMEM and then injected subcutaneously into SCID mice. Subsequently, animals were observed daily for tumor development. Three weeks after cell inoculation, tumors were isolated, and DNA was extracted by using proteinase K (Invitrogen) treatment. Mouse p53 mutant and wild-type alleles were amplified by PCR. Primers used and cycling conditions were as follows: 5′-TATACTCAGAGCCGGCCT-3′ (1), 5′-ACAGCGTGGTGGTACCTTAT-3′ (2), and 5′-TCCTCGTGCTTTACGGTATC-3′ (3), which specifically amplify the wild-type (470 bp, 1+2) or the mutant (600 bp, 1+3) mouse p53 allele, at 95°C for 30 seconds, 59°C for 30 seconds, and 72°C for 60 seconds for 30 cycles. PCR fragments were resolved on 1.2% ethidium bromide agarose gel. All experiments were performed at least 3 times, and similar results were obtained.
SA-β-gal staining of tumors
Mammary glands, breast tumors from MMTV-hN1IC mice, and xenografts of A549 or MDA-MB-231 human cancer cells were dissected, cut into approximately 5-mm-thick pieces, fixed in 1.25% glutaraldehyde, and subjected to standard SA-β-gal staining. 15 Tissue sections (4-µm-thick) from paraffin-embedded blocks were collected onto poly-L-lysine–coated slides and counterstained with eosin. Images shown were obtained by a Nikon COOLSCOPE II Digital Microscope (Tokyo, Japan).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: The study was supported from an EXCELLENCE-2011 grant (11EXC311) funded by the European Social Fund–European Union and Natural Resources.