
Abstract
Adaptor proteins are named for their function in assembling complexes of cellular proteins to execute and facilitate transmission of signals. The Crk family of adaptors consists of 2 members, Crk and CrkL. Crk, which was originally isolated as an oncogene, v-Crk, that transforms CEFs, has at least 2 splice variants, CrkI and CrkII, with differing biological activities. All Crk family proteins serve to act as molecular bridges between tyrosine kinases and their substrates and also modulate the specificity and stoichiometry of signaling processes. Signaling via CrkII and CrkL can be negatively regulated via tyrosine phosphorylation–mediated autoinhibition, while such a mechanism is not known to exist for CrkI. Although v-Crk clearly functions as a bona fide oncogene, in recent years, an emerging body of evidence suggests that cellular Crk proteins are overexpressed in human tumors and the expression levels correlate with aggressive and malignant behavior of cancer cells. These properties of Crk proteins make them potential cancer prognosis markers and therapeutic targets.
Introduction
Named for their ability to increase cellular tyrosine phosphorylation and transform primary chicken embryo fibroblasts (CEFs), CT10 regulator of kinase (Crk) is the prototypical member of a class of proteins called adaptors that contain Src homology-2 (SH2) and Src homology-3 (SH3) domains but lack intrinsic enzymatic activity. First discovered by Mayer and Hanafusa as the transforming gene product of an avian retrovirus, 1 the cellular homologs CrkI and CrkII 2,3 and the related Crk-like (CrkL) 4 are expressed in all tissues and identified across eukaryotic organisms. Over the past 2 decades, these ubiquitously expressed proteins have been implicated in complex and diverse physiological processes, ranging from regulation of the actin cytoskeleton in cell motility, phagocytic entry of apoptotic cells and pathogens into host cells, as well as cell cycle, apoptosis, and metabolism. 5,6 A general theme has also emerged that Crk proteins are dysregulated in several human malignancies, which will be the subject of this review.
There are several distinct members of the Crk family in addition to the retrovirus-encoded v-Crk. CrkI and CrkII were originally described as splice variants, 2 where CrkI resembled a structure analogous to v-Crk while CrkII possessed an extended C-terminus containing a proline-rich linker and an atypical SH3 domain (SH3C), which does not bind to classic proline-rich motifs 7,8 (Fig. 1). More recently, a third splice variant of crk, called CrkIII, has been described that is predicted to encode a protein with a truncated SH3C, 9 but the biological significance of this remains unknown. By contrast, CrkL has not been reported to be alternatively spliced. Despite the fact that CrkII and CrkL have a high degree of similarity, it is still not well understood how these isoforms vary in ligand affinities and specificity or how the 3-dimensional structures of CrkII and CrkL differ to engage key signaling partners. There are indeed expected to be differences in the signaling characteristics and/or tissue-specific expression of Crk and CrkL given the fact that the respective knockout mice have distinct phenotypes and that both proteins are required for embryonic development. 10,11
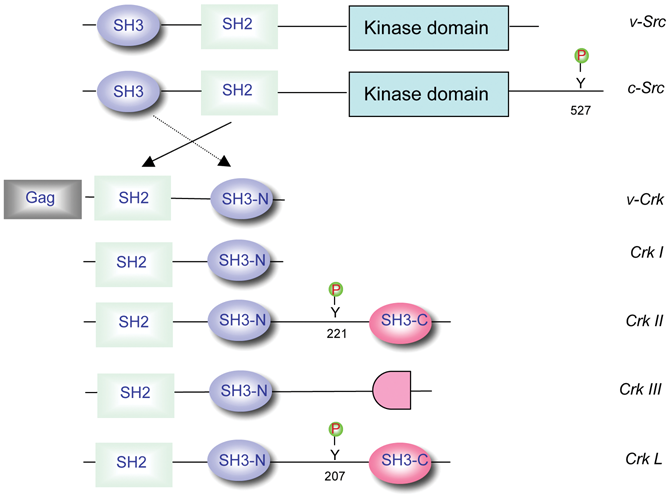
Schematic of the Crk family of adaptor proteins. Common protein interacting domains of the Crk proteins are indicated in comparison with Src. Gag refers to the group-specific antigen encoded by the CT10 avian retrovirus. Y527 in Src, Y221 in CrkII, and Y207 in CrkL are regulatory tyrosine motifs that bind intramolecularly to the SH2 domain.
Adaptors and Assembly of Signaling Complexes
The primary mechanism by which Crk and CrkL function is through the organized assembly of protein-protein complexes in a phosphotyrosine-dependent manner. 5 Shortly after the original publication describing Crk as an oncogene with sequence similarity to the N-terminal noncatalytic region of Src family kinases, Matsuda et al. reported for the first time that SH2 domains possess general phosphotyrosine binding activity 12 and that both the SH2 and SH3 domains were required for Crk signaling. 13 It was later realized that while the Crk SH2 domain recognized targets in a phosphotyrosine-dependent manner, specificity and high-affinity interactions were determined by amino acids carboxyl-terminal to the pTyr with the consensus pTyr-X-X-Pro. 14-16 A few years later, after several SH3N binding proteins were characterized, Knudson et al. used organized proline-oriented peptides to identify SH3 binding targets, and the consensus sequence for the Crk SH3N domain was determined to be Pro-x-x-Pro-x (Lys,Arg). 17 Subsequently, phage display screening technologies extended this paradigm of SH3 domain specificity, and several variations on the Pro-x-x-Pro/SH3 domain interactions emerged. 18,19 All Crk and CrkL binding partners, while diverse in their molecular function, utilize these common building blueprints to achieve a great degree of diversity in cell signaling. In contrast, the C-terminal SH3 domains of Crk and CrkL are atypical in that they do not bind polyproline type II (PPII)–containing motifs. 7 Indeed, the Crk SH3C domains are thought to confer allosteric regulation either on Crk or on other proteins. 20,21 This has raised the possibility that the atypical binding sites on Crk and CrkL SH3C may be involved in either lower affinity interactions or interactions that can be subjected to novel means of regulation.
Insight into the regulation of interactions between Crk and its ligands first emerged from biochemical studies 22,23 and then was confirmed by NMR 24 and X-ray crystallography. 20 These studies demonstrated that the biological activity of CrkII was controlled by tyrosine phosphorylation on Y221 by Abl (CrkL on Y207) that resulted in intramolecular autoinhibition. Such autoinhibition displaces the SH2 domain from binding phosphotyrosine motifs in trans. 20 This autoinhibition was notable for 2 important reasons: first, it clearly identified a mechanism by which v-Crk acquired transforming potential by deletion of the C-terminal–negative regulatory element; and second, it provided an analogous mechanism for the oncogenic activation of v-Crk as to the oncogenic activation of v-Src (deletion of Y527). 25,26 This pattern of activation of Crk and Src provided paradigms for the oncogenic activation of proto-oncogenes, a legacy established in the Hanafusa laboratory.
Crk in Human Cancers
Following the initial characterization of v-Crk as a transforming oncogene from the avian CT10 retrovirus, it became immediately clear that both v-Crk and CrkI possessed dominant oncogenic properties in chicken embryo fibroblasts, mouse NIH 3T3 cells, and when cells were injected subcutaneously into nude mice. Overexpression of CrkII 2 or CrkL 27,28 also transformed mammalian cells, although “weaker” in activity compared to CrkI mainly due to the negative regulatory tyrosine phosphorylation in their carboxyl-termini. However, forced CrkII expression recapitulates several features of malignant transformation, including alterations in focal adhesions, induction of Rac1-dependent membrane ruffles and lamellipodia, and altered adhesion. The original observations that a scaffolding protein such as v-Crk could transform cells implied that Crk was not simply a passive conduit for signaling but could amplify signaling output. In the last decade, the role of CrkII and CrkI in human cancers has steadfastly gained credibility. Both these proteins are frequently associated with aggressive and invasive human cancer phenotypes. Below, we review the evidence that Crk plays an important role in human cancer (Table 1).
Summary of the Analyses of Clinical Specimens for Crk and/or miR-126 Expression in Human Cancers
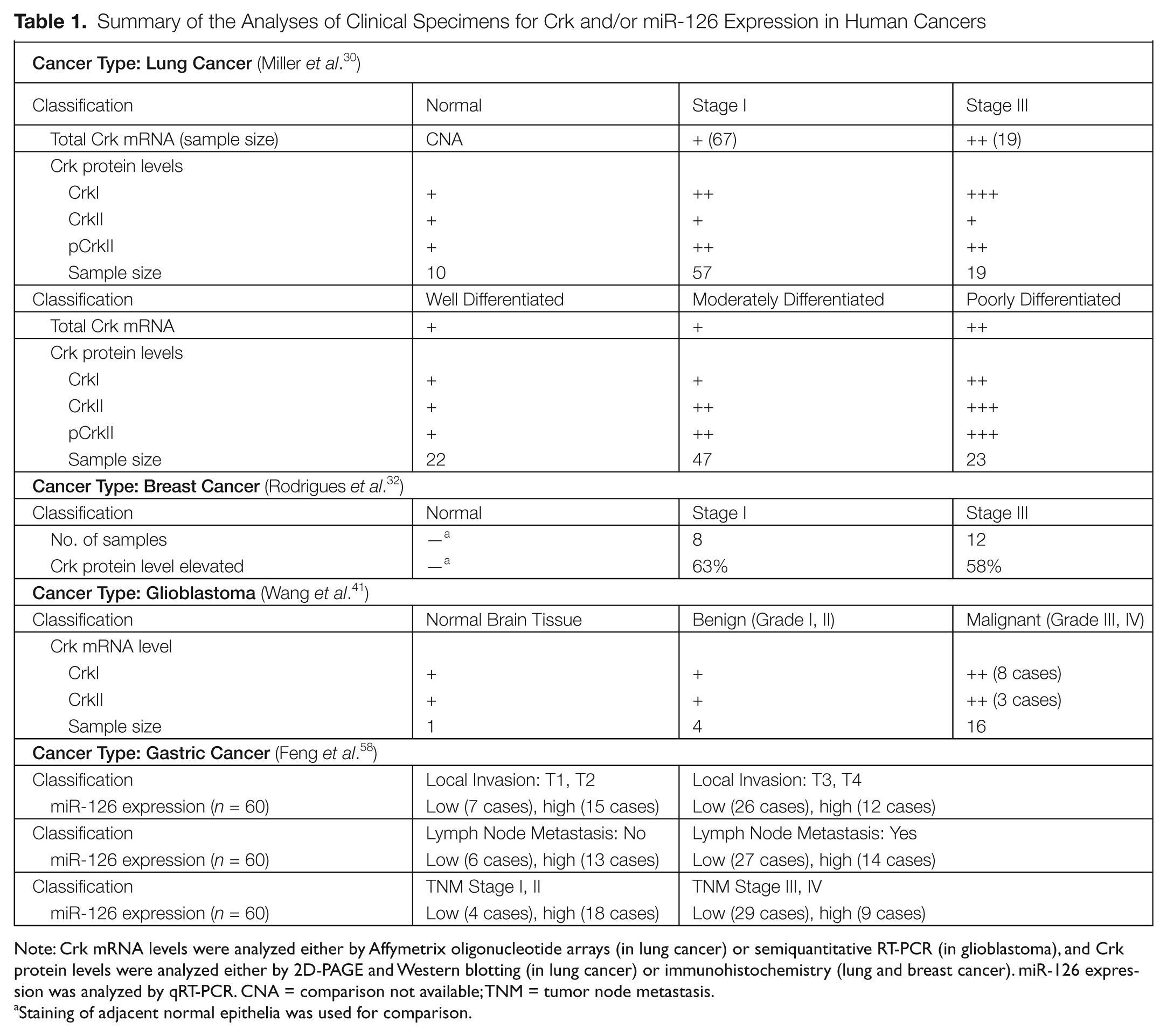
Note: Crk mRNA levels were analyzed either by Affymetrix oligonucleotide arrays (in lung cancer) or semiquantitative RT-PCR (in glioblastoma), and Crk protein levels were analyzed either by 2D-PAGE and Western blotting (in lung cancer) or immunohistochemistry (lung and breast cancer). miR-126 expression was analyzed by qRT-PCR. CNA = comparison not available; TNM = tumor node metastasis.
Staining of adjacent normal epithelia was used for comparison.
Lung Cancer
Lung cancer is the deadliest type of cancer for both men and women and is often diagnosed in a late incurable stage. 29 The vast majority of lung cancers are derived from carcinomas of epithelial cells, the 2 main types being small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), for which there are different treatment paradigms and expected survival outcomes. A study by Miller et al. was one of the first retrospective studies to profile Crk expression in a clinical cohort of well-described cancer patients. 30 Analysis of 86 lung adenocarcinomas (67 stage I and 19 stage III) and 10 uninvolved lung tissues for Crk mRNA expression by Affymetrix oligonucleotide arrays (Santa Clara, CA) revealed a significant increase in stage III compared to stage I and also in the more invasive tumors. In the same study, it was found that Crk mRNA expression was elevated in high-risk stage I and III tumors compared to low-risk stage I and III tumors. Moreover, poorly differentiated tumors expressed Crk at a significantly higher level than well or moderately differentiated tumors. Further, in 96 lung adenocarcinomas, CrkI and phospho (Y221)–CrkII protein levels were elevated in tumors versus normal lung tissue, while CrkI, CrkII, and phospho (Y221)–CrkII protein levels were significantly higher in poorly differentiated tumors compared to well-differentiated tumors, which correlates with poor survival outcomes.
More recently, Kim et al. reported on a study investigating genomic ampli-fications in NSCLC. 31 Using a cDNA microarray–based genomic profiling analysis of 128 lung cancer cell lines and tumor specimens (including adenocarcinomas and squamous cell carcinomas), CrkL amplification at the cytoband 22q11.21 was one of the most frequently amplified loci in lung cancer patients, which correlated with elevated CrkL expression in tumor cells. These investigators also showed that siRNA-mediated knockdown of CrkL in lung cancer blocked several hallmarks of aggressive tumor behavior, including cell cycle progression, survival, and motility as a measure of invasiveness. Further, CrkL overexpression in immortalized human bronchial epithelial cells significantly enhanced EGF-independent cell growth. 31
These studies also raise an interesting issue in relation to the interplay between Crk and CrkL in cancer cells as the aforementioned analyses suggest that both isoforms are overexpressed and contribute to the malignant behavior of the cells. Analogous to the Kim et al. study on CrkL in lung cancer alluded to above, Rodrigues et al. also showed that knockdown of Crk in the NSCLC cell line H1299 attenuated migration and invasion. 32 Clearly, an important future issue will be to resolve the individual roles of Crk and CrkL in tumor progression, as well as how they may crosstalk and/or synergize in driving aggressive phenotypes.
Breast Cancer
Analogous to overexpression of Crk in aggressive lung carcinomas, analysis of CrkII and CrkI in stage I and III breast tumors also revealed strong correlations between Crk expression and cancer staging in breast cancer. In the first study of its type, Rodrigues et al. performed immunohistochemical analysis of primary breast tumors (including 8 node-negative stage I and 12 node-positive stage III) and revealed that Crk (CrkI plus CrkII) protein levels were elevated in 60% of all tumors compared to adjacent normal ducts. 32 In the same study, it was found that siRNA-mediated knockdown of Crk significantly reduced invasion and migration of human breast cancer cell lines MDA-MB-231 and MDA-MB-435s.
Microarray analysis comparing T47D cells (a well-differentiated human breast cancer cell line) versus T47D cells overexpressing CrkII revealed that overexpression of CrkII resulted in the differential expression of a number of genes involved in processes important for tumor biology, namely, cell adhesion, migration, cytoskeletal reorganization, regulation of Rho GTPases, proliferation, and regulation of the cell cycle. Further, overexpression of CrkII promoted cellular dispersal and acquisition of a more mesenchymal phenotype characterized by the loss of adherens junctions reminiscent of epithelial-mesenchymal transition (EMT). 33 This study also corroborated previous molecular studies showing that Crk is involved in the breakdown of adherens junctions, a hallmark of EMT and required for the initial postreceptor responses to hepatocyte growth factor (HGF) in breast cancer T47D cells. 34 Finally, transgenic mice that express CrkII under the transcriptional control of the MMTV promoter show enhanced mammary gland branching with increased proliferation. Tumor incidence in these transgenic mice was significantly higher (17.6%) than in control mice (4%). 33
However, while the above scenario suggests that expression of Crk is a driver of motility and tumorigenicity in epithelial tumors of the breast, specific posttranslational modification of Crk, that is, phosphorylation on Tyr221, appears to underscore a distinct and unusual mechanism for tumor suppression. 35,36 For example, studies by Pasquale et al. showed that induction of apoptosis and suppression of migration of the human malignant breast cancer cell line MDA-MB-435c by EphrinB2, a ligand for EphB4 receptors, required phosphorylation of CrkII on Tyr221 by Abl, possibly disassembling Crk-mediated complexes including p130cas/Crk/DOCK180 and p130cas/Crk/C3G. 37 Crk Y221 phosphorylation by Abl was also found to be required for repression of MMP-2 expression upon EphrinB2 stimulation. 37 Treatment with EphrinB2 of tumors induced in mice (by injection of MDA-MB-435c cells) inhibited tumor growth, but this inhibition was abolished if EphrinB2 was administered along with imatinib, suggesting that negative regulation of Crk by Abl is essential for the antitumorigenic effects of EphrinB2. 37,38
Glioblastoma
Glioblastoma multiforme (GBM) is one of the most aggressive and invasive tumors in human cancer and has one of the highest mortality rates upon primary diagnoses. In WHO classifications of tumors of the central nervous system, GBM is graded and classified from grade I to grade IV, with the latter 2 stages (III and IV) considered malignant with poor survival outcomes. 39 Takino et al. reported on the expression of Crk in human glioblastoma by analyzing a small sample of human tissues and found that the expression of CrkI, but not CrkII, was elevated in several patients, which correlated with accelerated cell motility in a p130cas-, PI3 kinase–, and Rac1-dependent manner. 40 Subsequently, the Tanaka laboratory followed this study with a larger glioma sample and found that over 40% of malignant grade III and grade IV tumors had elevated CrkI but several had elevation in both CrkII and CrkI. 41 Analogous to the lung and breast, these studies showed that the malignant features of GBM cell lines could be defined by Crk expression, and siRNA-induced Crk knockdown decreased the motile behavior of cell lines and inhibited tumor growth in a mouse xenograft model. 41
Sarcomas
Synovial sarcoma is a rare malignant tumor of soft tissues that typically surround joints, tendons, muscles, blood vessels, and other fibrous tissues. It is particularly aggressive and metastatic and quite early becomes lymph node positive. In several sarcomas, including synovial sarcoma, amplified HGF/c-MET signaling has been implicated in tumor progression including EMT, tumor growth, and metastasis to the brain and lung. 42,43 Crk plays a central role in c-Met signaling 34 by binding to the tyrosine-phosphorylated scaffolding protein Gab1. 44 Binding of Crk to Gab1 sustains phosphorylation of Gab1 on Y307 that in turn induces activation of the Rac1 GTPase to promote cell migration (Fig. 2). Functionally, siRNA-mediated knockdown of Crk attenuated HGF-induced motility in human synovial sarcoma cell lines and also suppressed tumor formation in mouse xenograft studies. Subsequent studies further revealed that Crk promotes G1-S progression in synovial sarcoma cells mainly through the suppression of the CDK inhibitor p16 via Src-dependent activation of p38 MAPK. 45
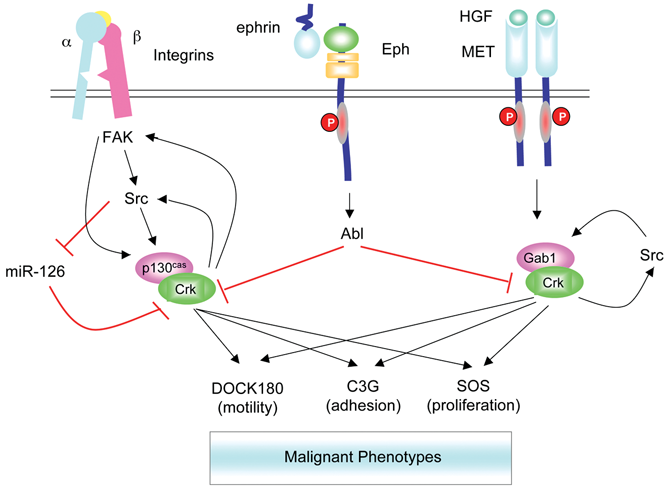
Feedback regulation of Crk and signaling pathways activated in malignant cells. Crk is engaged in multiple signaling pathways that drive the malignant behavior of cells. Tyrosine-phosphorylated p130cas (via integrins) and Gab1 (via Met receptor) act as molecular switches to assemble complexes of Crk with DOCK180, C3G, or Sos to stimulate indicated phenotypic outcomes. Elevation in Crk expression can also feedback and activate FAK and Src to further increase phosphorylation of p130cas in an amplification loop. Binding of Crk to tyrosine-phosphorylated Gab1 sustains phosphorylation on Y307 to possibly amplify downstream signaling. Activation of Src inhibits miR-126 defining another amplification loop. Finally, activation of Abl from upstream pathways including the engagement of Eph receptors can disassemble signaling complexes by inducing Crk phosphorylation on Y221. Similar pathways may operate for CrkL.
Ovarian Cancer
Analogous to the synovial sarcoma discussion above, ovarian cancer is another aggressive tumor type in which Crk may play an important role in disease progression. Most cases of ovarian cancer are detected only once the tumors are highly advanced, and often, these tumors proceed from initial to advanced metastatic disease in less than 1 year. Both CrkII and CrkI are expressed in primary ovarian epithelial cells and ovarian cancer cells and may actively contribute to the malignant phenotypes in these cells. 46 In studies by Linghu et al., siRNA knockdown of Crk suppressed growth rates, motility, tumor formation, and peritoneal invasion in a human mucinous cystadenocarcinoma (MCAS) cell line derived from an ovarian cancer patient. 46 Interestingly, these studies also showed that CrkL did not undergo compensatory up-regulation in Crk knockdown cells. Further studies showed that the Crk/DOCK180/Rac1 pathway also promoted malignant behavior of another ovarian cancer cell line SKOV3 and that knockdown of DOCK180 or ELMO1, but not C3G and Rap1, recapitulated these effects. 47 These data suggest that Crk is involved in the regulation of cell growth and motility during tumor progression in ovarian cancer.
Hematopoietic Cancers
The role of Crk and CrkL in hematopoietic malignancies remains understudied. CrkL, which has often been considered more highly expressed in myeloid- and lymphoid-derived cells, was shown to be involved to a greater extent than Crk in forming complexes with Bcr-Abl in transformed granulocytes and myeloid precursor cells. 48 Indeed, the interaction of CrkL with Bcr-Abl and its robust tyrosine phosphorylation on Tyr207 is often considered as a hallmark of Bcr-Abl activity and a prognostic indicator for remission in CML following imatinib treatment. 48 Indeed, both CrkL and Crk proteins, in the form of FRET reagents, have recently been employed to indirectly interrogate Bcr-Abl activity in CML and monitor drug resistance. 49,50
Despite the fact that Crk and CrkL both bind Bcr-Abl, it is still not clear what role these proteins play in leukemia. 51 Studies from several laboratories have shown that Bcr-Abl–mediated leukemogenesis continues unabated in CrkL (−/−) mice, 52,53 suggesting either redundancy with CrkI or CrkII or that CrkL signaling is not required for the manifestations of leukemia. 51 Very recently, the Druker laboratory has reported, however, that a Bcr-Abl mutant that collectively lacks the Grb2, Cbl, and CrkL binding sites fails to induce leukemia in mice in a bone marrow retroviral transduction model. 54 Hence, it still remains to be determined what role Crk plays in CML, particularly given reports that Crk can effect Abl kinase activity and signaling. 8,55
miR-126 Regulates Crk Expression
Recently, a mechanism that may explain how Crk becomes up-regulated in cancer cells has emerged in the form of identification of a micro-RNA (miR-126) that targets the Crk mRNA. Indeed, expression of miR-126 decreased Crk expression at the protein level and functionally resulted in the attenuation of malignant phenotypes of lung cancer cells by inhibiting invasion and migration. 56 Further studies showed that miR-126 was strongly suppressed in Src-transformed cells, offering a mechanism by which there is crosstalk between Src and Crk to influence tumor growth and invasion. 57 miR-126 was also identified in a screen of human gastric cancer patients to be significantly down modulated, 58 suggesting that miR-126 functions as a tumor suppressor in human gastric cancer. Finally, in the same study, miR-126 expression reduced Crk protein levels and inhibited cell invasion, migration, and metastasis in a mouse xenograft model.
Is Crk a Biomarker for Malignant Cancer Cells?
Do the above arguments and conclusions drawn from expression and signal transduction studies in a variety of human cancers imply that Crk and/or CrkL may be general biomarkers that predict aggressive tumor behavior? Early studies by Nishihara et al. evaluating human cancers showed that CrkII was significantly elevated in human lung and colon cancers, and typically expression correlated with lesion grade. 59 Similar findings are emerging with the assistance of iTRAQ-based mass spectrometry and microarray analysis, where Crk has been identified as a metastasis-associated biomarker in lymphatic and breast cancers. 60 More recently, Crk and CrkL were identified as “essential cancer causing genes” using genome-wide shRNA library screening platforms combined with computational approaches from 12 cancer cell lines representing diverse cancer types. 61 These data are in agreement with the general idea that Crk and/or associated signaling components may be potential prognosis markers and therapeutic targets.
Mechanisms and Interventions
Although a general pattern of Crk overexpression correlating with the malignant features of cancer cells has already emerged, extending the expression and experimental analyses of Crk to a much larger set of cancer cell lines and cancer tissues will permit a more systematic assessment of the extent to which Crk is overexpressed and its role in human cancers. Efforts to categorize Crk signaling in relation to specific cancer phenotypes will eventually lead to an evaluation of Crk and/or associated signaling components as therapeutic targets. Given the centrality of Crk in the aforementioned invasive tumor phenotypes, it is conceivable that introduction of miR-126 or shRNA to Crk into tumor cells might have therapeutic value in attenuating metastasis of invasive tumors. Proof of concept experiments in mouse models will provide an estimate of the likelihood of success of these strategies.
Further studies are also required to dissect out the exact mechanisms by which Crk becomes up-regulated in human cancer and how it drives aggressive phenotypes. There is a large body of evidence on investigating the latter, as complexes of either p130Cas/Crk/DOCK180 or Gab1/Crk/DOCK180 lead directly to Rac1 activation, altered actin cytoskeletal assemblages, and motility via interactions with WAVE and Arp2/3 complexes 5 (Fig. 2). Crk can also bind to C3G via its SH3N domain, and the resultant p130Cas/Crk/C3G complexes can activate Rap1-inducing inside to outside signaling, leading to increased integrin activation, p130Cas phosphorylation, and enhancement of cell motility. 62 It is noteworthy that the above pathways can be amplified by positive feedback loops established by Crk up-regulation, such as what is detectable in various human cancers. Forced expression of CrkII results in enhanced activation of FAK that further increases p130Cas and paxillin phosphorylation 14 or enhanced Gab1 phosphorylation via Src, 47 thereby enhancing cell motility. Such amplification loops may not only enhance motility but can activate Src, 45 which in turn may down-regulate miR-126 and increase Crk levels 57 in a second amplification loop.
In a recent study to elucidate which signaling pathways of Crk mediate cell growth, Mayer et al. employed a systematic approach in CrkI-transformed NIH3T3 cells by individually knocking down SH3N binding proteins, including DOCK180, C3G, SOS1, and Abl/Arg. 63 RNAi-mediated knockdown of C3G or SOS1 suppressed anchorage-independent growth and tumor formation in nude mice, while knockdown of DOCK180 had little effect on tumor growth. These data suggest that motility (DOCK180-dependent) and tumorigenicity (SOS1- and C3G-dependent) are separable in the NIH3T3 model, and such systematic knockdowns in other cancer cells would add important insight into which pathways contribute to specific phenotypes.
Role of Abl in Crk Signaling
While Abl is the sole tyrosine kinase that interacts with Crk SH3N, its role in Crk signaling during tumor progression remains surprisingly elusive. On one hand, in the Mayer study, 63 knockdown of Abl in CrkI-transformed NIH 3T3 cells resulted in enhanced anchorage-independent growth and tumorigenic potential. Such observations are consistent with studies by the Pasquale laboratory, 37 showing that Abl mediates the antitumorigenic effects of EphrinB2 in breast cancer by negatively regulating CrkII-mediated signaling. On the other hand, equally compelling evidence also suggests that Abl is activated downstream of deregulated EGFR and Her2/ErbB2 in aggressive breast cancer lines and enhances the invasiveness of those cell lines. 64,65 However, reconciliation of this role of Abl with that of mediating the antitumorigenic effects of EphrinB2 by phosphorylation of CrkII at Y221 is not obvious as both tumor-suppressive EphrinB2/EphB4 signaling 37 and mitogenic/invasive EGF/EGFR signaling have been shown to induce phosphorylation of CrkII at Y221. 66 Conceivably, Crk may play a yet unrealized role in either or both signaling pathways that impinges on the balance between EphrinB4 and EGF signaling in breast cancer.
Special Remarks
The authors and editors dedicate this review to Hidesaburo Hanafusa, head of the Laboratory of Viral Oncology at Rockefeller University from 1973 to 2000. While initially dedicated to the biology of Src in the 1970s and 1980s, much of the effort in the Hanafusa laboratory shifted away from Src and toward Crk after the discovery of v-Crk in 1988. As fate would permit, conceptual advances on Src and other retroviral oncogenes studied by Hanafusa paved, in a large way, the road to unraveling the function of Crk in cell transformation. Besides obvious similarities in the composition of modular SH2 and SH3 domains in Crk and Src, both genes were similarly “hijacked” by RNA tumor viruses, and both exhibited C-terminal truncations that resulted in the loss of SH2-pTyr–mediated negative regulatory elements. These mechanisms remain today as textbook explanations for oncogene activation and the oncogene theory of human cancer.
The emerging importance of Crk in a vast variety of human cancers is reminiscent of the history of Src and the revelation that Src is important in human cancer. In this respect, Src was not always appreciated as a relevant player in human cancer. In the early 1990s, it was still not convincing whether Src had a significant role in human cancer since it was rarely mutated and not at all on Tyr527 in tumor samples. Of course, the evidence for a role of Src in human cancers is now quite compelling, and Src kinase inhibitors are entering the mainstream of clinical cancer therapy. 67 The story of Crk in cancer therapeutics remains to be completed, although it is now clear to play a central role in malignant transformation.
Footnotes
Acknowledgements
The authors thank Dr. Charles Reichman for helpful discussion and dedicate this review to the late Dr. Hidesaburo Hanafusa.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This paper was in part supported by a grant from the National Institutes of Health [R01 GM080308].