
Abstract
The Crk family of adaptor proteins (CrkI, CrkII, and CrkL), originally discovered as the oncogene fusion product, v-Crk, of the CT10 chicken retrovirus, lacks catalytic activity but engages with multiple signaling pathways through their SH2 and SH3 domains. Crk proteins link upstream tyrosine kinase and integrin-dependent signals to downstream effectors, acting as adaptors in diverse signaling pathways and cellular processes. Crk proteins are now recognized to play a role in the malignancy of many human cancers, stimulating renewed interest in their mechanism of action in cancer progression. The contribution of Crk signaling to malignancy has been predominantly studied in fibroblasts and in hematopoietic models and more recently in epithelial models. A mechanistic understanding of Crk proteins in cancer progression in vivo is still poorly understood in part due to the highly pleiotropic nature of Crk signaling. Recent advances in the structural organization of Crk domains, new roles in kinase regulation, and increased knowledge of the mechanisms and frequency of Crk overexpression in human cancers have provided an incentive for further study in in vivo models. An understanding of the mechanisms through which Crk proteins act as oncogenic drivers could have important implications in therapeutic targeting.
Introduction
Crk proteins were first discovered in the late 1980s as the oncogene fusion product, v-Crk, of the CT10 avian sarcoma virus. 1 Unlike known viral oncogenes at that time, such as v-Src and v-Abl, v-Crk lacks catalytic activity. 1 Instead, v-Crk is composed of the viral gag sequence followed by a region of sequence similarity with nonreceptor tyrosine kinases. 1 This region of homology was later understood to contain a Src homology 2 (SH2) domain and a SH3 domain, which bind to phosphorylated tyrosine and proline-rich motifs, respectively.2,3 Overexpression of v-Crk was sufficient to induce the transformation of chicken and rodent fibroblasts and promote tumors with short latency, 1 which required both an intact SH2 and SH3 domain, 4 highlighting the potential involvement of cellular Crk proteins in tumorigenesis. Increased cellular tyrosine phosphorylation was identified as a hallmark of v-Crk–transformed cells, which suggested abnormal cellular kinase activity and led to the name Crk, or CT10 regulator of kinase.1,4 Matsuda et al. 5 identified that in transformed cells, v-Crk stably associated with tyrosine-phosphorylated proteins. This required an intact SH2 domain and led to the understanding that the v-Crk SH2 domain, and subsequently SH2 domains in general, binds phosphotyrosine residues.
Cellular homologs of v-Crk include the CRK gene (chromosome 17p13), which is alternatively spliced to give rise to 2 well-characterized proteins (CrkI and CrkII) 6 as well as a third splice variant (CrkIII) 7 , and a second gene CRKL, or Crk-like (chromosome 22q11). 8 CrkI resembles v-Crk and is composed of 1 SH2 domain and 1 SH3 domain. CrkII contains a linker region and an atypical C-terminal SH3 domain, while CrkIII is predicated to encode a protein with a truncated C-terminal SH3 domain (Fig. 1A). CrkL shares 60% overall homology with CrkII, with increased homology in the SH2 and SH3 domains. 8 The SH2 domain of Crk proteins binds to phosphorylated tyrosine motifs with an optimal consensus sequence of pTyr-x-x-Pro. 2 The N-terminal SH3 domain (SH3N) binds proline-rich motifs of the polyproline II (PPII) subtype with the consensus Pro-x-x-Pro-x-(Lys,Arg), 3 whereas the C-terminal SH3 (SH3C) domain is atypical and does not bind to conventional PPII motifs. 9 The CrkI, CrkII, and CrkL proteins are expressed in all embryonic tissues tested and in adult murine tissues to varying degrees.10,11 CrkL expression is highest in adult hematopoietic tissues and low in many epithelial tissues, whereas Crk exhibits the highest expression in the brain, lung, and kidney and low expression in bone marrow. 10
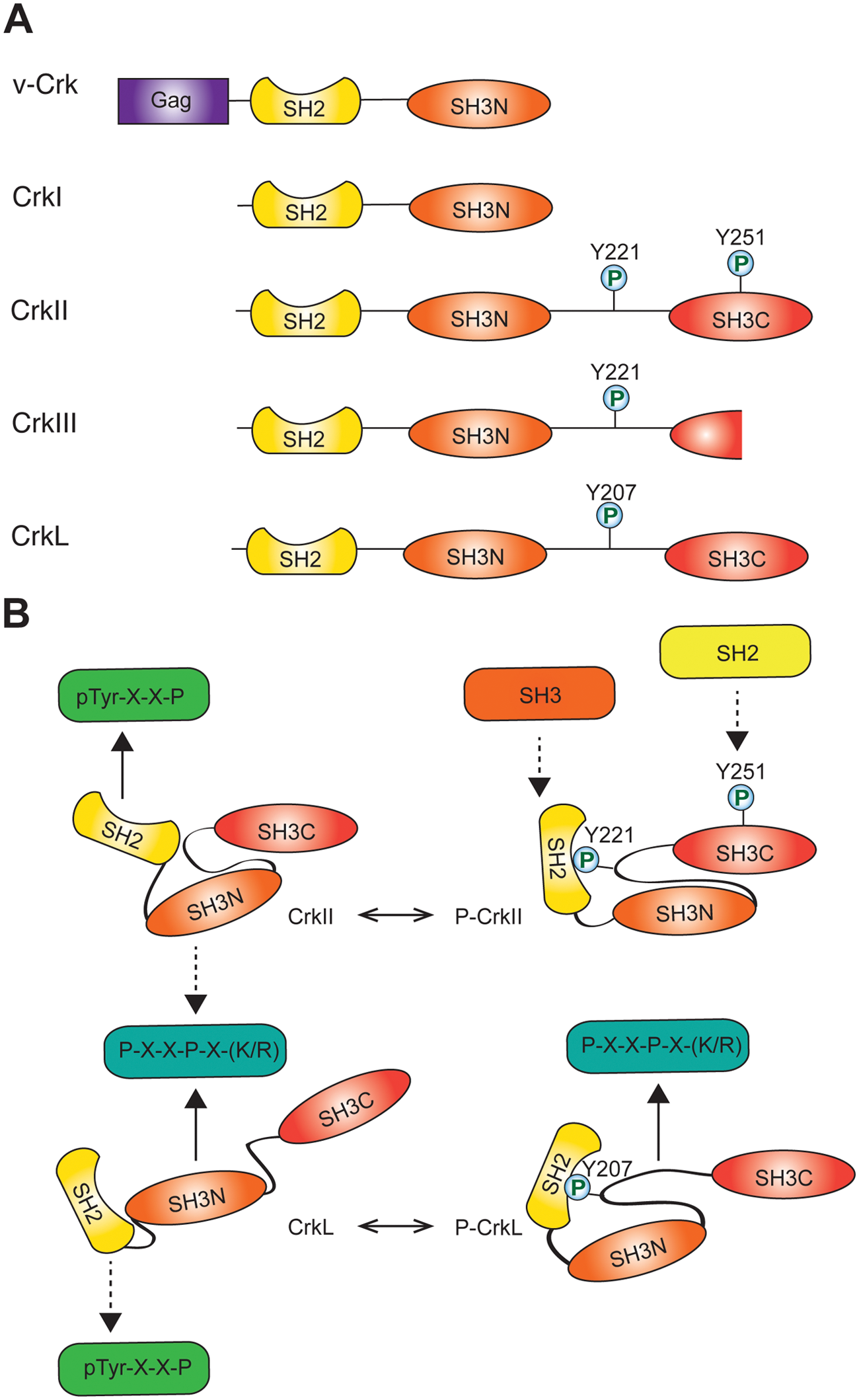
Crk family of adaptor proteins. (
Distinct Developmental Functions of Crk Proteins
Both CRK and CRKL genes are contained in chromosomal regions identified in human deletion syndromes, highlighting the importance of these genes in developmental processes. The CRK gene is localized within the 17p13.3 region deleted in Miller-Dieker syndrome. 12 Miller-Dieker syndrome is characterized by facial dysmorphisms, impaired cognitive functioning, and growth restriction. 13 Mice lacking CrkII are viable and phenotypically normal; however, ablation of both CrkI and CrkII results in embryonic lethality with defects in cardiac and craniofacial development.14,15 CRKL is located in the 22q11 region commonly deleted in patients with DiGeorge/velocardiofacial syndrome (DGS/VCFS), which occurs in humans with a frequency of 1 in 4000 live births. 16 The developmental defects observed in DGS/VCFS involve neural crest cells, and phenotypic hallmarks include aortic arch malformations, craniofacial dysmorphisms, and thymic abnormalties. 17 CrkL – / – mice die in utero by embryonic day 16.5, exhibiting multiple cranial and cardiac abnormalities consistent with defects in neural crest cells, 18 although the severity of these defects appears to be strain dependent. 19 The similarity between the phenotype of Crk-null mice and the corresponding human deletion syndromes highlights an important and conserved role for Crk during development and supports the applicability of mouse models for studying Crk in cancer. The embryonic lethality observed in both Crk – / – mice and in CrkL – / – mice indicates unique roles for each Crk gene product and a lack of functional redundancy during development. Tissue-specific deletion of Crk proteins using Cre-loxP recombination to circumvent embryonic lethality has identified functions for Crk proteins in neuronal migration in the developing murine brain and in foot process effacement in models of glomerular disease.20,21
Regulation of Crk Proteins
Overexpression of CrkI, CrkII, or CrkL is sufficient to promote morphological transformation of fibroblasts, and overexpression of CrkI or CrkL also supports adhesion-independent growth.6,22 CrkI, which resembles v-Crk, has the highest transforming activity in fibroblasts, promoting high levels of phosphotyrosine and the capacity of these cells to induce tumors in nude mice. 6 Hence, although structurally similar, this suggested the differential regulation or function of Crk proteins in tumorigenesis.
The increased transforming activity of CrkI and v-Crk compared with CrkII and CrkL has been attributed to the absence of an autoinhibitory mechanism, whereby the SH2 domain of CrkII and CrkL forms an intramolecular interaction with a phosphorylated tyrosine (Y221 and Y207, respectively) in the SH3N-SH3C linker region of each protein (Fig. 1B).23,24 Intramolecular binding decreases the availability of the CrkII/L SH2 domain for other tyrosine-phosphorylated binding partners. This provides a mechanism for oncogenic activation of v-Crk by removal of the linker region containing the phosphorylated tyrosine residue.
Despite a high degree of homology in their functional domains, CrkII and CrkL are organized with distinct structural architectures (Fig. 1B). 25 In contrast to the relatively open conformation of CrkL, the unphosphorylated form of CrkII adopts a compact structure in which protein binding to the SH3N domain is partially occluded by the inter-SH3 linker region, and the autoinhibitory structure of phosphorylated CrkII further decreases SH3N accessibility (Fig. 1B). 26 This autoregulatory mechanism is mirrored in the chicken CrkII protein by cis-trans proline isomerization (Pro238) in which the favored cis form results in hydrophobic contacts between the CrkII SH3N and SH3C domains to regulate SH3N binding. 27 Thus, although mechanisms of CrkII regulation are not conserved across species, both chicken and human CrkII proteins exhibit autoregulation by the Crk C-terminal linker and SH3 domain, demonstrating an evolutionary importance of maintaining regulation of Crk adaptor function. While these regulatory mechanisms decrease availability of the CrkII SH3N for binding, the CrkL SH3N maintains ligand affinity in both the phosphorylated and unphosphorylated protein conformations. 28 Unphosphorylated CrkL is also subject to autoinhibitory regulation by polar contacts between the SH2 and SH3N domains that result in partial occlusion of the SH2 domain. 28 Therefore, structural variability in linker and SH3C domains confers relative increased affinity of CrkII and CrkL for their SH2 and SH3 domain binding partners, respectively. These structural differences could confer both specific regulation and the ability of Crk proteins to differentially engage with both upstream and downstream signaling proteins, resulting in independent functional activities.29,30
Regulation of autoinhibition of CrkII and CrkL through tyrosine phosphorylation, and hence biological activity, is mediated under physiological and pathophysiological conditions by the Abl tyrosine kinase. 31 In addition to promoting an autoinhibited conformation, phosphorylation of CrkII on tyrosine 221 and intramolecular binding by the CrkII SH2 domain expose a proline-rich motif in the SH2 domain that binds the Abl SH3. 32 In addition to phosphorylation on Y221, recent evidence supports that Abl can also phosphorylate Y251 in the CrkII SH3C domain, creating a potential binding site for additional SH2 domain–containing proteins. 33 Hence, this potentially provides a mechanism for phosphorylation-regulated recruitment of SH3 and SH2 domain–containing proteins to CrkII, which may result in differential localization in response to biological stimuli.
Crk proteins are also predominant phosphorylation substrates for the Bcr-Abl fusion oncogene, which is found in greater than 95% of chronic myelogenous leukemias (CML) and 20% to 30% of acute lymphoblastic leukemias (ALL). 34 Although CrkII and CrkL share a high degree of homology within their functional domains, CrkL is the major tyrosine-phosphorylated protein in Bcr-Abl–driven CML patient neutrophils. 34 The preferential binding of Bcr-Abl to CrkL, even in the presence of CrkII, suggests disparity in interaction properties and differential regulation of Crk proteins by Bcr-Abl or Abl tyrosine kinases. Within the last 5 years, advances in the understanding of Crk protein conformations have provided an explanation for the different binding affinities of these 2 very similar proteins. 25
Crk Proteins in Oncogenic Signaling
The Crk protein family, a prototype for the study of adaptor proteins, has contributed much to our understanding of protein complex formation in signal transduction. Although early studies focused on Crk proteins in the transformation of fibroblasts, the study of Crk proteins in epithelial cell models has further elucidated oncogenic signaling by Crk (Fig. 2). For example, overexpression of CrkI/II or CrkL in normal epithelial cells, or in cancerous epithelial cells of breast, lung, oral, or head and neck origin, enhances epithelial-to-mesenchymal transitions (EMT), characterized by the breakdown of adherens junctions, enhanced cell dispersal, and remodeling of cortical actin.30,35-37 Conversely, RNA interference targeting the CRKI/II gene decreases the migration, invasion, and tumorigenesis of human cancer cells including breast, ovarian, and oral squamous cell carcinoma cell lines, implicating Crk as a critical signal transducer for cell invasion in many human cancers.35,37-39
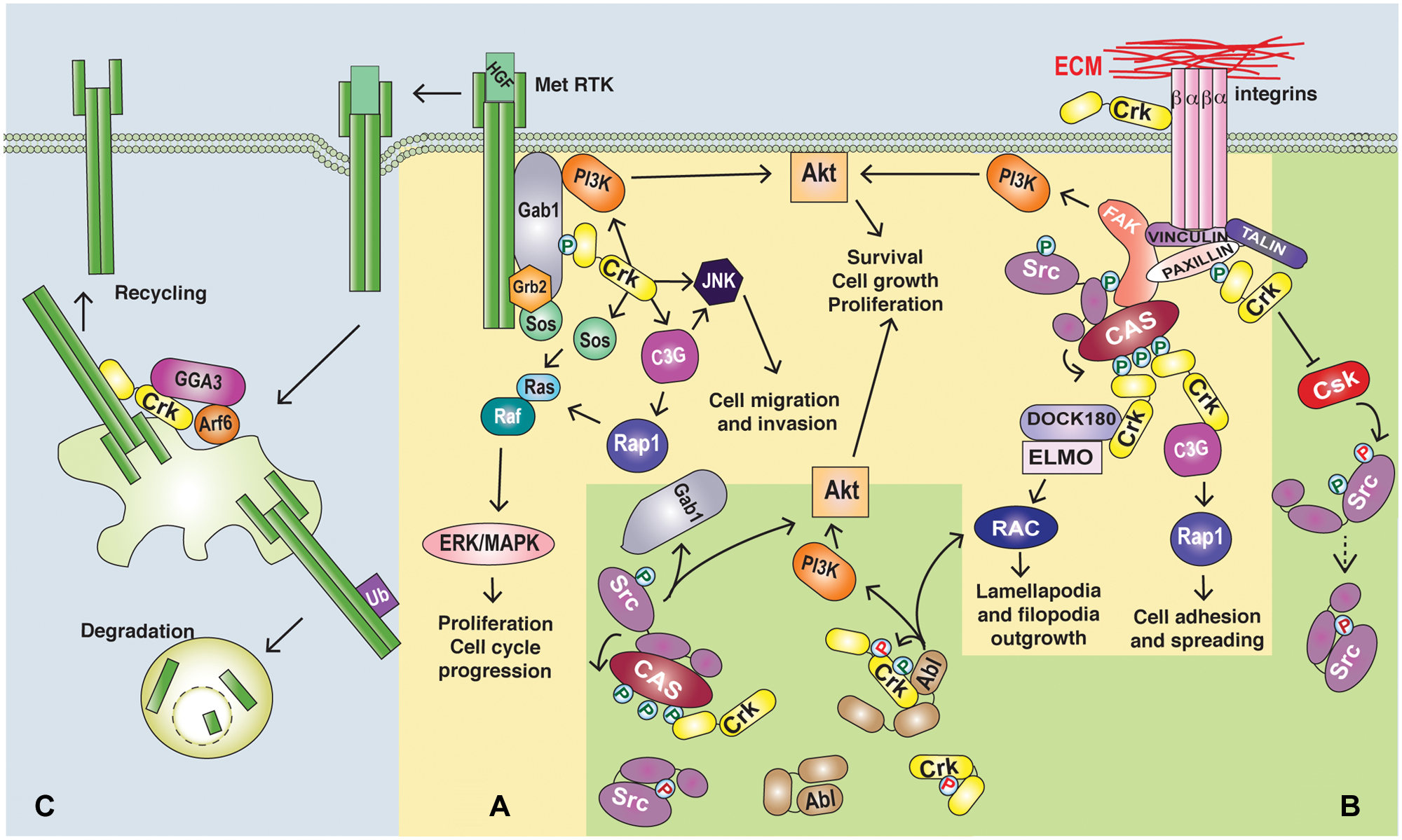
Roles of Crk proteins in oncogenic signaling. (
The ability of Crk proteins to induce the transformation of fibroblasts and EMT-like phenotypes in epithelia supports a role for Crk proteins in the amplification and/or initiation of oncogenic signals. Consistent with this, Crk proteins act in signaling cascades that regulate a variety of cellular processes through A) protein complex formation downstream from receptor activation, B) regulation of cellular tyrosine kinase activity, and C) upstream roles in signal initiation (Fig. 2 and Table 1).
Roles of Crk Proteins in Oncogenic Signaling
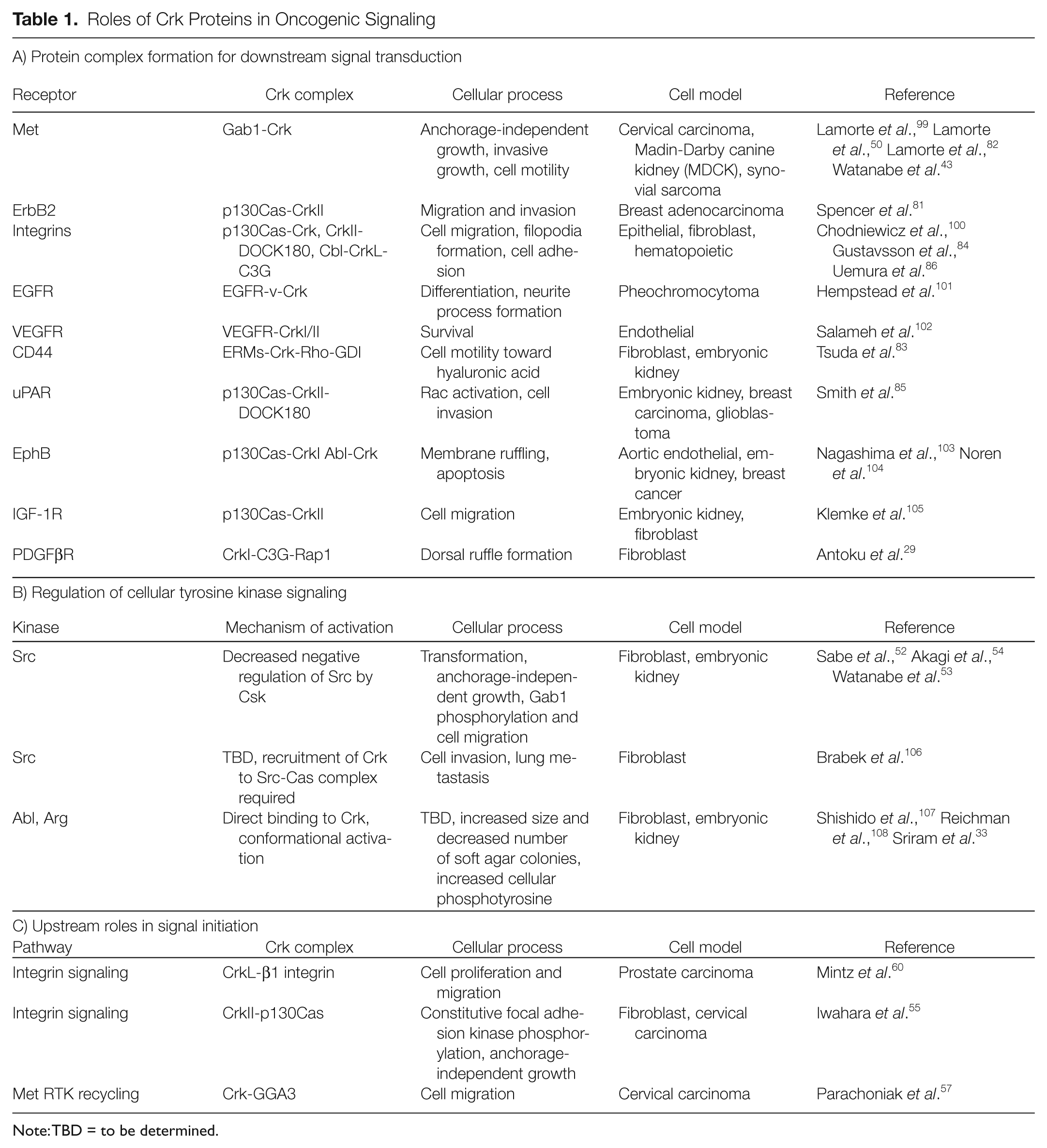
Note: TBD = to be determined.
Protein complex formation in signal transduction
Crk adaptor proteins are believed to act as a molecular bridge by recruiting downstream effectors to upstream phosphorylated tyrosine motifs. Within this context, Crk protein complex formation is predominantly induced downstream from oncogenic tyrosine kinases as well as integrin engagement. 40 Multiple binding partners have been identified for both the SH2 and SH3 domains of Crk, illustrating diverse roles for Crk in cellular processes. 41 The best-characterized SH2 domain binding partners are the tyrosine-phosphorylated scaffold proteins p130Cas and paxillin, which are effectors downstream of integrin activation,2,42 as well as the scaffold proteins Gab1 and Gab2, which are effectors downstream from many tyrosine kinases.43,44 Crk SH3N domains bind to diverse proteins including the nonreceptor tyrosine kinases c-Abl and Arg, which themselves phosphorylate Crk proteins (Y221 and Y251 in CrkII; Y207 in CrkL), regulating autoinhibition.31,45 The SH3N of Crk also associates with a proline-rich motif in the stress-activated protein kinase JNK and has a critical role in JNK activation downstream of growth factor stimulation. 46 Major Crk SH3N domain binding proteins include the guanine nucleotide exchange factor proteins, SOS1, 47 C3G, 3 and DOCK180, 48 leading to the activation of Ras, Rap1, and Rac1, 41 allowing Crk to couple diverse upstream signals, spatially and temporally, with the regulation of small GTPases. An essential function for SOS1 was identified for the tumor formation of CrkI-transformed fibroblasts, whereas either C3G or SOS1 was important for anchorage-independent growth or morphological transformation in vitro (C3G). 49 Similarly, specific recruitment of Crk to the Gab1 scaffold downstream from the Met/HGF receptor tyrosine kinase in epithelial cells is required for the efficient activation of Rac1 and Rap1 and essential for a program of invasive growth. 50 The diverse roles of Crk proteins in different signaling cascades suggest that Crk overexpression can cooperate with a variety of oncogenic signals modulating transformation and make Crk proteins therapeutic targets owing to their central downstream signaling role.
Regulation of cellular protein tyrosine kinases by Crk
Elevated cellular phosphotyrosine levels as a result of Crk overexpression may also be mediated through Crk regulation of protein tyrosine kinase activity. Multiple studies identify Abl and Src as candidate kinases for aberrant activation by overexpression of Crk proteins. Abl promotes Crk autoinhibition by phosphorylating Tyr221 (CrkII) and Tyr207 (CrkL) and also phosphorylates Tyr251 in the atypical CrkII SH3C domain (Fig. 1B). 33 This creates a potential binding site for the Abl SH2 domain, providing a mechanism to enhance the activation of Abl by relieving intramolecular binding to the kinase domain. 33 The Src tyrosine kinase is also subject to intramolecular autoinhibitory interactions engaging both Src SH2 and SH3 domains. 51 Although the relationship between Src and Crk has yet to be fully understood, elevated levels of Crk may positively regulate Src by displacing Csk, a negative regulator of Src, from Src signaling complexes.52,53 Crk mediated transformation induces the increased activity of Src family kinases and constitutive phosphorylation of p130Cas and FAK.54,55 Similarly, Crk overexpression promotes Src-dependent tyrosine phosphorylation of the Gab1 scaffold protein, recruitment of Crk, and enhanced focal adhesion turnover and cell migration. 53 Interestingly, a recently proposed model for Gab1 structure56 indicates that binding of proteins such as Crk could alter Gab1 three dimensional conformation to allow additional protein-protein interactions and signal transduction. Gab1 contains multiple Crk binding sites that appear to be clustered in a potential 3-dimensional structural domain involving intramolecular interaction with its PH domain. Crk engagement with this scaffold may open its conformation 56 and impact its capacity to undergo other protein-protein interactions.
Upstream roles for Crk in signaling cascades
A role for Crk as an upstream regulator of cell signaling is also consistent with observations that Crk proteins amplify or initiate signaling pathways for transformation. A role for Crk upstream of the Met receptor tyrosine kinase has recently been identified, with Crk involvement in Met recycling back to the plasma membrane after internalization. 57 This prolongs signaling by the Met RTK and enhances Met-dependent cell migration. 57 Upstream roles for Crk proteins have also been identified in integrin signaling. Crk proteins activate Rap1 through recruitment of the C3G exchange factor to sites of Crk localization such as phosphorylated p130CAS, which in turn can enhance integrin affinity for ligands by an inside-out mechanism.58,59 An extracellular role for CrkL in binding to the β1 integrin regulatory plexin-semaphorin-integrin (PSI) domain has also been associated with enhanced integrin-mediated cell proliferation and migration, 60 highlighting new functions for Crk proteins in cancers.
Crk Proteins in Human Cancer
Since their discovery, Crk proteins have emerged in a wide variety of human cancers. 61 While mutations in the functional domains or regulatory phosphorylation sites of Crk genes have not been detected, elevated expression of both CrkI/II and CrkL is found in several human cancers including breast, ovarian, oral, stomach, pancreatic, lung, and glioblastomas.37,61,62 Where studied, increased Crk protein levels have been associated with advanced-stage cancers with high-grade and enhanced proliferation.37,38,63,64 One mechanism for the dysregulation of CrkL protein expression has emerged, where CRKL is localized within a region of genomic amplification in non–small cell lung cancer (NSCLC). 65 A posttranscriptional mechanism resulting in elevation of CrkI and CrkII protein levels was also revealed when Crk was identified as a target of the tumor suppressor microRNA miR-126, which is implicated in the regulation of processes important for angiogenesis, inflammation, and cancer progression. 66 Decreased miR-126 has been observed in multiple human cancers compared to normal tissue, including breast, prostate, gastric, colorectal, and pancreatic tumors,62,66-68 and is associated with elevated Crk protein levels in gastric and pancreatic tumor tissues.62,69 Taken together, these studies show the elevation of Crk proteins in diverse human cancers and an association with an aggressive tumor phenotype. While this does not determine a causative role for Crk in cancer initiation or progression, the use of relevant cell-based and mouse models is beginning to identify a requirement for Crk proteins in tumor progression.
Transgenic mice expressing multiple CrkL transgene copies each under the control of the CRKL promoter were generated to study the contribution of CrkL to tumorigenesis. 70 Interestingly, CrkL transgenic mice developed tumors of diverse types and tissues of origin with low penetrance and long average latency. Tumors observed included lung adenoma, mammary adenocarcinoma, lymphoma, and fibrosarcoma and appeared with latency ranging from 8 to 26 months. 70 This supports a role for CrkL in tumorigenesis but suggests that additional events are necessary for tumor onset.
Hematopoietic cancers
Bcr-Abl proteins, which result from a translocation involving chromosomes 9 and 22, are found in over 90% of CML and have been proven to have a causal role in leukemia development using transgenic mouse models.71,72 The demonstration that CrkL was the major tyrosine-phosphorylated protein in neutrophils from patients with CML containing the Bcr-Abl fusion kinase led to the establishment of initial murine models of Crk proteins in cancer for the study of CrkL in hematopoietic disease. 34 Importantly, CrkL is not detectably phosphorylated in peripheral blood cells lacking Bcr-Abl, demonstrating that phosphorylation of CrkL in CML is aberrant and could indicate a specific role for CrkL in pathogenesis. 73 Importantly, reduction in tyrosine phosphorylation of CrkL has prognostic value, monitoring for the response to imatinib, a small molecule inhibitor that targets Bcr-Abl kinases and is used clinically to treat CML. 74
The consistent association of abnormal CrkL phosphorylation with Bcr-Abl activity is also true in leukemic tissues of Bcr-Abl transgenic mice, indicating that transgenic mice are useful models for studying the role of Crk proteins in hematopoietic disease. 10 When crossed with transgenic mice expressing P190 BCR-ABL under the control of the mouse metallothionein-1 promoter, 70 CrkL overexpression shortened the latency for the development of B-cell lineage lymphoblastic leukemia/lymphoma induced by Bcr-Abl, decreasing the average age at death from 413 days to 300 days. This demonstrates that P190 Bcr-Abl and CrkL overexpression are additive in disease progression and supports a role for CrkL involvement in pathogenic signaling from Bcr-Abl. However, crossing a viable strain of CrkL – / – mice with the P190 BCR-ABL model did not alter the development of B-cell lineage leukemia/lymphoma, suggesting that CrkL may not be essential for Bcr-Abl–driven B-cell lineage leukemias. 19 However, fetal liver cells from an embryonic lethal CrkL-null mouse model showed a dosage-sensitive requirement for CrkL in P210 Bcr-Abl–induced transformation. 75 Hence, it is possible that the requirement for CrkL differs with genetic background or with the breakpoint in the Bcr-Abl fusion protein, as P210 Bcr-Abl and P190 Bcr-Abl promote different leukemia phenotypes of primarily myeloid and lymphoid lineages, respectively.71,76
Lung cancers
Increased expression of CRKI/II mRNA in lung adenocarcinoma is associated with poorly differentiated tumors and poor survival. 64 Elevated CrkI/II expression in NSCLC could be due to a recurrent genomic deletion of the 9q34.3 region containing miR-126, which targets the CRKI/II gene, 77 whereas overexpression of CrkL in NSCLC is associated with a recurrent 22q11.21 amplicon containing the CRKL gene (Fig. 3).65,78 Expression of miR-126 is also epigenetically silenced in NSCLC cell lines and primary NSCLC, providing another mechanism for increased CrkI/II expression. 79 High levels of CrkL are also observed in NSCLC cell lines lacking amplification of 22q11.21, suggesting that other mechanisms can contribute to overexpression. 78 Importantly, shRNA-mediated suppression of CrkL in NSCLC cells decreased proliferation, suggesting that, when overexpressed, CrkL functions as a required oncogene in these cells. 78 Further support for a key functional role for CrkL in NSCLC was provided when CRKL was identified as the top scoring gene in an shRNA-based screen for essential genes in cancer cell proliferation. 80 CrkL may have important implications for patient treatment, as CrkL overexpression has been linked to resistance to the EGFR inhibitor gefitinib in NSCLC cell lines and patient samples. 78
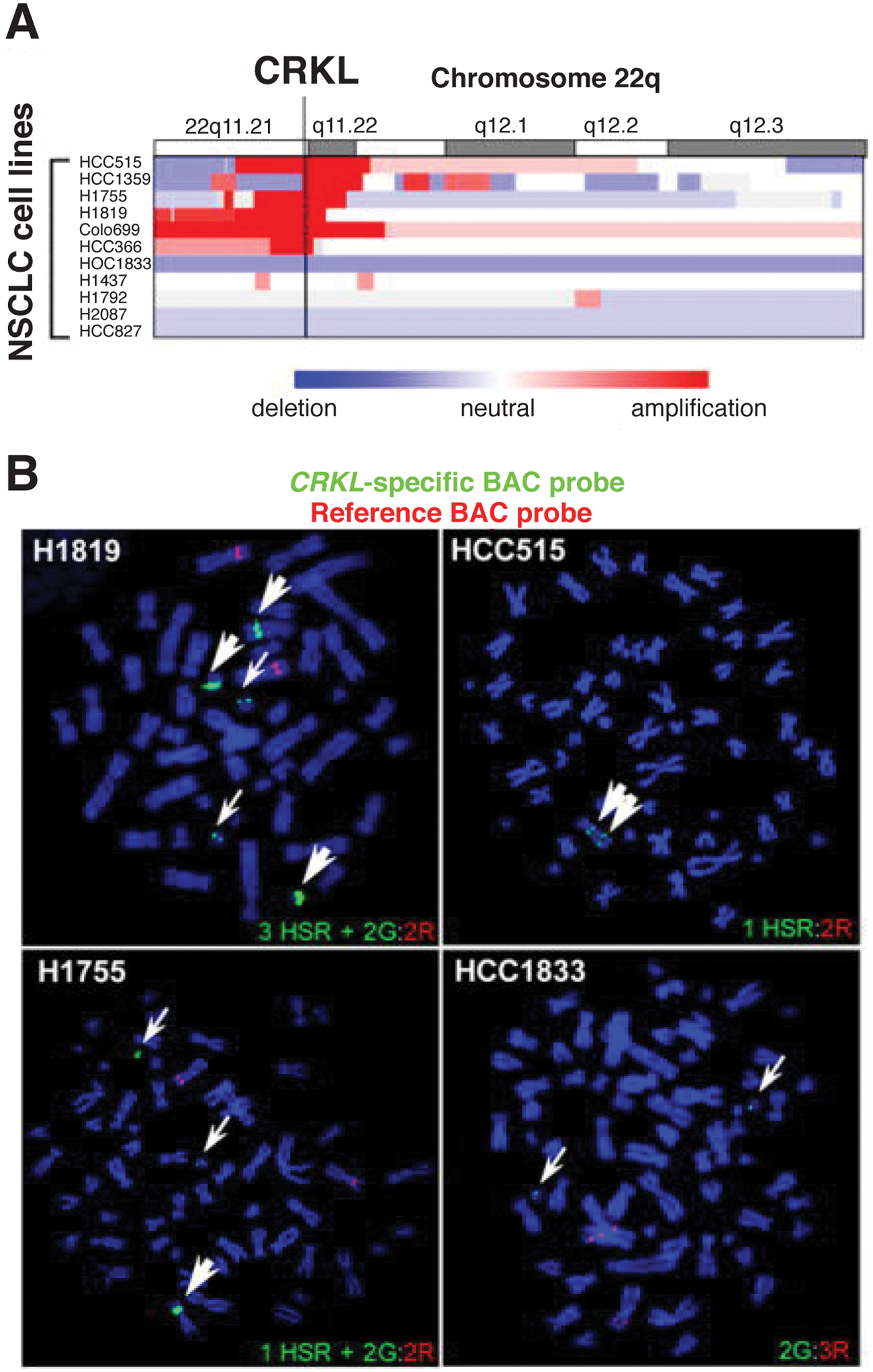
Amplification of CRKL in non–small cell lung cancer (NSCLC) cell lines.
78
(
Despite identification of CrkL in primary NSCLC, in vivo tumorigenesis studies have been restricted to xenotransplant models of NSCLC cell lines. 78 A subcutaneous injection model of established NSCLC cell lines showed that doxycycline-inducible knockdown of CrkL caused the arrest of tumor growth and increased apoptosis. 78 Although CrkL in lung cancer has not been specifically studied in a transgenic mouse model, lung adenoma was among the tumors observed in transgenic mice overexpressing multiple copies of CrkL under its endogenous promoter, but a causal role for CrkL in lung tumorigenesis cannot be concluded from the number of mice studied. 70 The increasing evidence indicating that CrkL is both aberrantly expressed and functionally important in NSCLC warrants further mechanistic studies of roles for CrkL in initiation, progression, and resistance to treatment.
Breast cancers
Crk adaptor proteins mediate signaling downstream from several upstream signals implicated in breast cancer progression, including EGFR, Met, CD44, ErbB2, and integrins.81-86 A role for Crk in promoting breast tumorigenesis is suggested by multiple studies observing elevated Crk protein expression in breast tumors compared with adjacent normal ducts.35,87 Breast cancer can be divided into luminal, HER2-positive, and basal-like molecular subtypes by gene expression profiling. 88 Therapies targeting estrogen and HER2 receptors have shown efficacy in luminal and HER2-amplified breast cancers, but basal-like cancers are predominantly negative for estrogen, progesterone, and HER2 receptors, called triple negative, and are therefore associated with poor prognosis due to the lack of targeted therapeutics. An understanding of Crk proteins in breast cancer must differentiate between the different subtypes, as changes in cellular signaling may alter both the pathways in which Crk proteins act and the amount of reliance on Crk for promoting oncogenic signaling. A gene expression signature generated following overexpression of CrkII in a luminal breast cancer cell line, when applied to gene expression datasets of breast cancers, associates with high-grade tumors and basal subtypes. 38 This suggests that Crk-enhanced signaling promotes changes, such as EMT, that are associated with basal breast cancers. 89 Patients with a high correlation with the Crk signature exhibit a significant decrease in disease-free survival, 38 suggesting that Crk proteins may play a role in breast cancer disease progression, highlighting a need for a better mechanistic understanding of Crk signaling in breast cancer.
The use of tissue microarrays of human breast tumors containing representative samples from breast cancer subtypes (n = 254) revealed that elevated levels of CrkI/II and CrkL proteins are associated with high-grade tumors consistent with the association of the Crk gene expression signature with grade. 38 Importantly, within high-grade, triple-negative breast tumors, increased levels of Crk proteins significantly correlated with increased cell proliferation, suggesting that independent of tumor grade, Crk proteins are important in the proliferation of basal breast cancer cells (Fig. 4). Consistent with this, knockdown of all 3 Crk proteins by RNA interference in basal breast cancer cell models decreased in vivo proliferation but not apoptosis and abrogated orthotopic tumor outgrowth in a mammary fat pad injection model. 38 Similarly, re-expression of the Crk-targeting miR-126 in metastatic breast cancer cell lines suppressed tumor growth and decreased proliferation without affecting apoptosis. 90 These studies demonstrate that Crk proteins are associated with the progression of basal breast cancer.
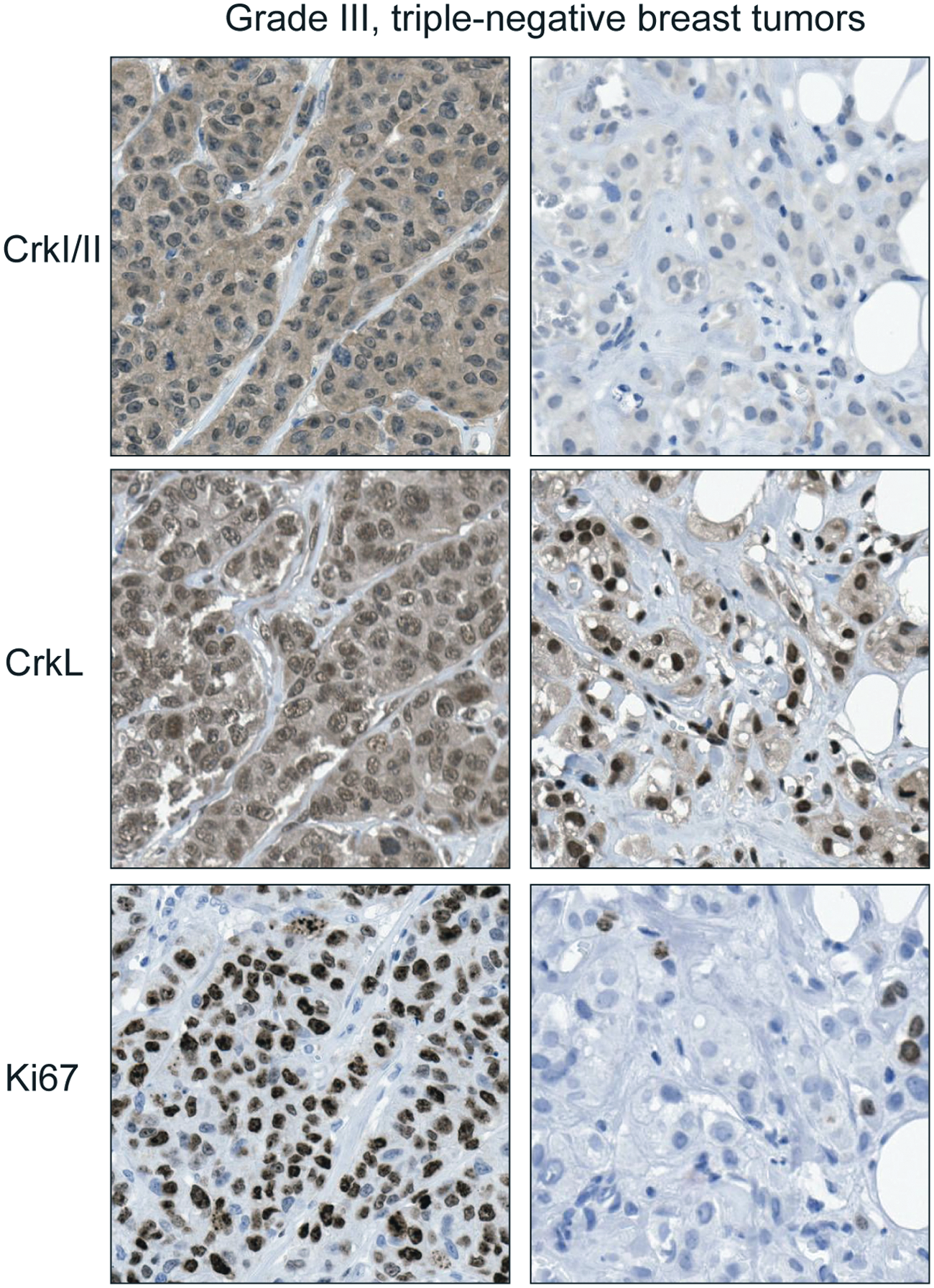
Expression of Crk adaptor proteins is associated with an increased proliferative index in human triple-negative breast tumors. Immunohistochemistry using antibodies to Crk, CrkL, and the proliferation marker Ki67 in triple-negative, grade III breast tumors. 38 Panels on left show a tumor with high Crk protein and proliferative index. Panels on right show a tumor with low Crk protein and proliferative index.
The possibility of a causative role for Crk proteins in breast tumorigenesis was examined through the generation of transgenic lines expressing CrkII in the mammary gland under the control of the mouse mammary tumor virus (MMTV) promoter. 91 Consistent with roles for Crk in development, pubertal MMTV-CrkII mice exhibited delayed ductal outgrowth throughout the mammary fat pad and increased number and size of terminal end bud structures. 91 Mammary glands of postpubertal mice showed enhanced branching and cell proliferation, which has been associated with predisposition to tumorigenesis in other models. 92 MMTV-CrkII mice developed mammary tumors with low penetrance (17.6%), long latency (~445 days), and diverse phenotypes. 91 The presentation of the MMTV-CrkII model is consistent with requirements for cooperating oncogenic events to initiate tumorigenesis, suggesting that overexpression of CrkII in the mammary gland may enhance the response to oncogenic signaling but is not transforming alone. Elucidating the role of Crk in breast cancer requires further study of Crk-accelerated tumorigenesis in combination with suitable mouse models of breast cancer, with attention paid to the subtype of human breast cancer recapitulated by each model. 93
Although molecular mechanisms of how Crk proteins contribute to the growth of basal breast cancer have yet to be determined, Crk proteins have been shown to be sufficient and required for the elevated tyrosine phosphorylation of p130Cas in xenotransplant models of basal breast cancer cell lines. 38 Furthermore, immunofluorescence performed on frozen human breast tumors shows a consistent correlation between increased Crk protein and p130Cas tyrosine phosphorylation. 38 Interestingly, a transgenic mouse model of p130Cas under the control of the MMTV promoter shows similarities to the MMTV-CrkII mouse model.91,92
Future Perspectives
After more than 20 years of study, new roles and complexities of Crk signaling continue to be discovered. As discussed in this review, Crk family proteins are now recognized as important modulators of cell proliferation, migration, and invasion downstream from many upstream signals, and elevation of Crk proteins is associated with multiple human cancers. As high expression of Crk proteins is required for their transforming activity, and is now functionally associated in several human cancers with high grade and poor outcome, a more detailed screening of Crk family expression, subcellular localization, and chromosomal amplification in additional human cancers should be undertaken. A more comprehensive understanding of mechanisms of control of gene expression, promoter regulation, and epigenetic modulation is required. Identification of exogenous regulators for Crk gene expression under physiological and pathophysiological conditions is also important. New discoveries of mechanisms of regulation of Crk protein structure and function require further understanding. For example, the recent observation that serine phosphorylation of CrkII plays a role regulating cell motility in NSCLC cells 36 and the finding that an amino-terminal fragment of CrkI/II acts in endoplasmic reticulum stress–induced signaling contributing to the activation of mitochondrial proapoptotic machinery warrant further mechanistic investigation. 94 These newly identified roles for Crk proteins illustrate both the diversity of cellular processes in which Crk acts as well as the likelihood that there are other undiscovered roles for Crk proteins.
The observations that elevated Crk proteins enhance the activation of Src, Abl, as well as FAK kinases in experimental conditions should be pursued in human cancers and could identify new subsets of patients that benefit from targeted therapeutics to these kinases. This could be particularly interesting in the context of breast cancer in which Abl expression is associated with aggressive disease,95,96 and Crk expression correlates with proliferation of the poor prognosis basal subtype. 38
The development of peptides that bind with high specificity to the Crk and CrkL N-terminal SH3 domains and disrupt complex formation with SH3 binding partners provides a mechanism through which to study the effects of inhibition of Crk proteins on tumorigenesis.97,98 Peptides with specificity for CrkII versus CrkL, or for specific Crk protein complex formation, 75 may prove useful in targeting specific subsets of cancers.
The characterization of CrkL as an oncogenic driver in NSCLC, along with advances in understanding of the structural regulation and the mechanisms of overexpression of Crk proteins, has provided a solid foundation for a more extensive study of Crk in in vivo models. As knowledge of Crk in different signaling pathways grows, it will be important to determine what the key Crk signaling events are in different cancers and the impact of Crk overexpression on patient response to different therapeutics.
Footnotes
Acknowledgements
The authors thank Dr. William Hahn for the opportunity to reproduce his data in this review. Thanks to Andrea Lai and Richard Vaillancourt for helpful comments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: Work from the authors’ laboratory is funded by grants from the Terry Fox New Frontiers program project (#020002) and from the Canadian Institutes of Health Research (#IOP-92594) to M.P. E.S.B. is supported by a studentship from the Department of Defense (#BC100836). M.P. holds the Diane and Sal Guerrera Chair in Cancer Genetics at McGill University.