
Abstract
The efficient management of misfolded protein aggregates is essential for cell viability and requires 3 interconnected pathways: the molecular chaperone machinery that assists protein folding, the proteasome pathway that degrades misfolded proteins, and the aggresomal pathway that sequesters and delivers toxic protein aggregates to autophagy for clearance. Although autophagy is generally considered as nonselective degradative machinery, growing evidence supports the existence of a selective autophagy that specifically targets protein aggregates for clearance. This “quality control autophagy” is established by specific ubiquitin E3 ligases, autophagic substrate ubiquitination, and specific ubiquitin-binding proteins p62 and HDAC6. In this context, quality control autophagy is similar to the proteasome system and utilizes ubiquitin tags for substrate recognition and processing. Here, I will discuss the recent progress toward understanding the molecular basis of this unique form of ubiquitin-dependent autophagy in protein aggregate clearance and its relevance to disease.
Introduction
Misfolded proteins are the inevitable by-products of biogenesis, resulting from genetic mutations, inappropriate protein complex assembly, aberrant modifications, and environmental stress. It is estimated that up to 30% of newly synthesized proteins are not folded properly. 1 More than simply being nonfunctional, misfolded proteins are prone to form aggregates that can interfere with normal cellular function. Thus, misfolded proteins and aggregates are closely monitored, processed, and eliminated to prevent their accumulation in cells. 2 This protein quality control machinery works at multiple levels. It augments the proper tertiary structure of unfolded or misfolded proteins via molecular chaperones including heat shock proteins (Hsp) and promotes the degradation of aberrant proteins that fail to refold. 3 It is generally thought that misfolded proteins are recognized and polyubiquitinated by specific ubiquitin E3 ligases. This polyubiquitination then targets misfolded proteins to proteasomes for degradation. 3 The ubiquitin-dependent degradation of misfolded proteins by proteasomes constitutes a critical part of the cytoprotective quality control machinery. Defects in ubiquitin-proteasome machinery lead to the accumulation of misfolded proteins and aggregates, a condition commonly found in neurodegenerative disease.
Once aggregated, misfolded proteins are not degraded efficiently by proteasomes. 4 Oligomeric or aggregated proteins simply cannot pass through the narrow barrels of proteasomes where polypeptides are digested. In fact, accumulation of protein aggregates can inhibit proteasome activities by clogging the proteasomes or other indirect mechanisms, resulting in further accumulation of misfolded proteins that aggregate. 4,5 The transgenic expression of aggregation-prone proteins, such as mutant androgen receptor with expanded polyglutamine (poly Q) stretches, markedly inhibits proteasome activities in a Drosophila neurodegenerative model, suggesting that this feed-forward production of protein aggregates is a major contributing factor to neurodegeneration. 6 The machinery responsible for the clearance of toxic protein aggregates faces 2 basic challenges: first, it must “recognize” protein aggregates, and second, its degradative component must be able to physically “accommodate” the large size of protein aggregates. These challenges are solved by an intricate network that integrates ubiquitin modification into protein aggregates to provide a specific recognition code, which are read by ubiquitin-binding factors equipped to coordinate the concentration and delivery of protein aggregates to a versatile degradative system–lysosome-based autophagy.
The Aggresome: a Collection Center for Protein Aggregates
Inclusion bodies have long been recognized as collections of misfolded proteins and aggregates. Their prevalence in almost all forms of neurodegenerative disease has stimulated intense research interest to unravel the pathophysiological significance of these protein junkyards. 7 The identification of a microtubule-organizing center (MTOC)–localized inclusion body, the aggresome, has provided a critical model to understand how toxic protein aggregates are managed. A simple and elegant aggresome model was first proposed by Kopito et al., whereby cytoplasmic protein aggregates are collected by dynein motors and transported along the microtubule network to the MTOC, where they are concentrated and become the aggresome. 8 Aggresomes and Lewy bodies, the hallmark cytoplasmic inclusion bodies found in neurons affected by Parkinson disease, share striking biochemical and molecular characteristics. 9 Accordingly, the formation of Lewy body–like inclusion bodies is an active cellular response to the accumulation of protein aggregates in neurons. The formation of aggresomes likely serves two functions: 1) to prevent widespread intracellular toxicity originated from protein aggregates by actively removing them from the cytoplasm and concentrating them to aggresomes and 2) to permit efficient degradation of these toxic aggregates by MTOC-targeted proteolytic apparatus, including autophagy. 2
Autophagy: A Versatile Lysosome-Based Degradative System
Autophagy was initially discovered as an unusual cellular response to nutrient starvation where cells undergo “self-digestion” of their own cytosolic contents in order to recycle essential macromolecules for survival. In the most characterized form of autophagy, macroautophagy, starvation induces a unique double-membrane structure that sequesters cytosolic contents into autophagic vesicles and forms the autophagosome. Autophagosomes subsequently fuse to lysosomes where the autophagic contents are degraded by lysosomal enzymes. 10 Autophagy essentially provides a vehicle to deliver cytosolic contents to lysosomes. Electron microscopic images of autophagic vesicles accumulated in yeast mutant strain deficient in vacuole (equivalent of lysosomes) protease activity revealed the presence of large organelles, including ribosomes and mitochondria. 11 This observation highlights the remarkable ability of the autophagosome to capture cellular contents and organelles of various sizes. This feature makes autophagy an ideal solution for disposing of protein aggregates, which cannot be processed by proteasomes. Indeed, protein aggregates have long been observed inside the autophagosomes induced by proteasome inhibition. 12 The molecular nature of this protein aggregate–associated autophagy, however, has just recently been uncovered.
Autophagy has been generally characterized as a nonselective degradative pathway activated by nutrient starvation. This is logical considering that the primary purpose of autophagy is to efficiently replenish macromolecules, and consequently, all cytoplasmic residents could be potential substrates. It has been, however, long recognized that autophagy is not solely dedicated to nutrient recycling. One main function of this poorly defined nutrient-independent “basal” autophagy is to enforce intracellular quality control by disposing of protein aggregates and damaged or supernumerary organelles that are toxic or no longer needed. 10 In contrast to the classic starvation-induced autophagy, this form of autophagy, referred to as quality control (QC) autophagy, must have the ability to distinguish aberrant protein aggregates and damaged organelles from their normal counterparts. The characterization of two ubiquitin-binding proteins, p62 and HDAC6, has revealed that, similar to proteasome-mediated protein degradation, a ubiquitin-dependent system is also responsible for conferring the substrate specificity for QC autophagy.
p62: A Receptor for Ubiquitinated Protein Aggregates
The formation of autophagosomes requires the conjugation of yeast ATG8-related proteins (LC3-A, B, C and GABARPA, GABARPAL-1, L2, L3 in mammals) to the lipid phosphatidylethanolamine, which results in autophagic membrane expansion and fusion. 13,14 Interestingly, ATG8-related family members are structurally related to ubiquitin. 15,16 Similar to ubiquitin conjugation, ATG8-related molecules require specific E1-activating and E2-conjugating enzymes to form linkage to phosphatidylethanolamine via the C-terminal glycine. 13 Remarkably, ATG8-related molecules are not only required for autophagosome formation but also serve as a physical link to ubiquitinated protein aggregates.
p62, also known as SQSTM1, is a common component of the ubiquitin-positive inclusion bodies found in neurodegenerative and liver disease as well as in cultured cells subjected to misfolded protein stresses. 17 p62 contains multiple protein-protein interactive domains. It binds ubiquitinated proteins via its UBA domain and can undergo oligomerization through its PB1 domains. 18 Accordingly, overexpression of p62 in cells can bring ubiquitinated proteins together to form aggregates. 18,19 This activity is highly relevant as the formation of aggresome-like inclusion bodies is significantly impaired in p62-deficient cells. 20 Similarly, large ubiquitin-positive protein aggregates, which accumulate prominently in autophagy-deficient neurons or hepatocytes in tissue-specific ATG7 knockout mice, do not form if p62 is genetically ablated. 21 Thus, p62 is required for the concentration of ubiquitinated protein aggregates. The second crucial function of p62 in protein aggregate clearance was discovered when an electron microscopy (EM) study revealed that a portion of p62 is concentrated to vesicles with a double membrane, a defining feature of autophagosomes. 19 Remarkably, in a search for p62 protein interactors, LC3, the essential component of the autophagic membrane, was identified. 20,21 A reverse screen using LC3 as the bait identified p62 as an interactor. These findings led to a simple model whereby p62 acts as a bridge that connects ubiquitinated protein aggregates to autophagosomes. A similar activity was recently reported for a p62-related protein, NBR-1. 22 This model explains how autophagosomes are specifically targeted to protein aggregates (Fig. 1).
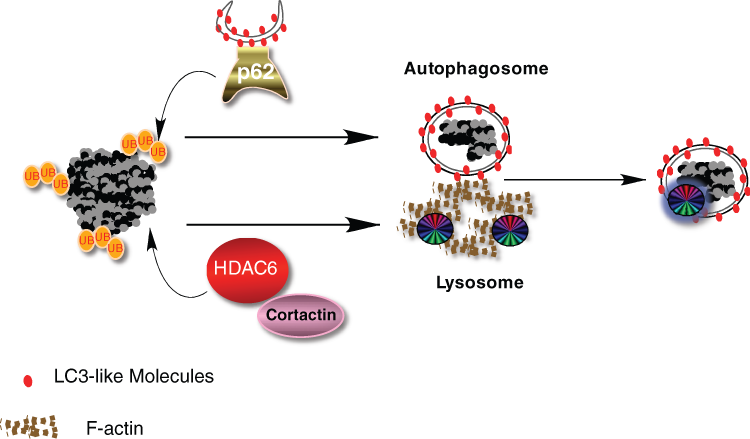
Ubiquitin modification on protein aggregates recruits ubiquitin-binding factors p62 and HDAC6. Specific ubiquitin (ub) chains associated with protein aggregates recruit p62, which in turn brings in autophagosomes by its binding to LC3-like molecules on the autophagosomal membrane. Ubiquitin also recruits HDAC6, which promotes cortactin-dependent F-actin remodeling and subsequent autophagosome-lysosome fusion.
HDAC6: A Ubiquitin-Binding Protein Deacetylase that Controls Aggresome Formation and Autophagosome Maturation
The protein deacetylase HDAC6 is the second crucial component required for protein aggregate clearance. Although most HDAC members are involved in chromatin remodeling and gene transcription, HDAC6 is uniquely localized to the cytoplasm, contains a ubiquitin-binding domain (BUZ finger), and associates with both the microtubule and actin cytoskeleton. 23-28 Its ability to bind ubiquitinated misfolded proteins and dynein motors led to the proposal that HDAC6 facilitates the transport of protein aggregates to the MTOC and forms the aggresome. 27 In HDAC6-deficient cells, large aggresomes do not form; instead, dispersed microaggregates are observed throughout the cytoplasm indicative of a failure to transport protein aggregates to the MTOC. 27 Similar ubiquitinated protein aggregates are observed in neurons in HDAC6 knockout mice. 29 This phenotype, however, is not identical to that of p62-deficient cells, where no protein microaggregates are observed. 20,21 The 2 different phenotypes suggest that p62 might act upstream to HDAC6 whereby p62 concentrates misfolded protein aggregates, and HDAC6 directs their dynein-dependent transport to the MTOC. In this model, p62 and HDAC6 work in tandem to concentrate protein aggregates to the aggresome-like inclusion bodies.
In HDAC6-deficient cells, protein aggregate clearance is severely impaired, indicating a defect in autophagy. However, HDAC6 is neither required for autophagy activation nor autophagosome targeting to protein aggregates. Autophagy consists of 2 discrete steps: the formation of double-membrane autophagosomes that sequester away cytosolic constituents, and the delivery of autophagic substrates to lysosomes by fusion of these two compartments. As autophagosomes lack intrinsic enzymatic activity, their fusion to lysosomes represents a crucial step for productive autophagy. HDAC6 is specifically required for this final step of autophagy and promotes autophagosome-lysosome fusion by recruiting cortactin-dependent actin remodeling machinery. 29 By activating cortactin via deacetylation, 30 HDAC6 promotes the formation of an F-actin network that stimulates autophagosome-lysosome fusion. 29
These findings show that ubiquitination controls autophagy at two different levels: the recruitment of autophagosomes to protein aggregates via p62, and the efficient fusion of autophagosome to lysosomes via HDAC6-dependent actin remodeling. This arrangement ensures the specificity and efficiency for disposing of ubiquitinated protein aggregates. These unique properties suggest a simple model wherein HDAC6 and p62 independently recognize and bind specific ubiquitin moieties that mark protein aggregates and facilitate their clearance (Fig. 1). In this context, specific ubiquitin modification serves to nucleate and assemble autophagic factors with ubiquitin-binding activities. This arrangement is reminiscent of the ubiquitin-dependent assembly of DNA repair machinery at DNA damage foci, where specific ubiquitination events recruit repair proteins with ubiquitin-binding activities. 31 In both cases, ubiquitin plays a central role in providing both specificity and efficiency.
Ubiquitin-Binding p97/VCP in QC Autophagy
Biochemical purification of HDAC6 has revealed that endogenous HDAC6 resides in a protein complex containing p97 and PLAP, the vertebrate homology of yeast Cdc48 and UFD3, respectively. 23 p97, which is also called valosin-containing protein (VCP), is a member of the AAA-ATPase family. p97/VCP interacts with various ubiquitin-binding partners to regulate multiple ubiquitin-dependent processes. UFD3, also a known Cdc48 interactor, is a ubiquitin-binding protein whose specific function is less understood but can affect the levels of the free ubiquitin pool. 32 Mutations in p97/VCP cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD), a disease characterized by protein aggregate accumulation, neurodegeneration, and muscle defects. 33 Interestingly, the expression of disease-causing 97/VCP mutants inhibits autophagosome-lysosome fusion, a phenotype similarly observed in HDAC6-deficient cells. Thus, an auto-phagosome maturation defect could contribute to the development of IBMPFD. 34,35 In both yeast and mammalian cells, Cdc48 or p97/VCP is also required for aggresome formation. 36,37 It is likely that HDAC6 works in conjunction with p97/VCP to control auto-phagosome maturation and aggresome formation in a ubiquitin-dependent mechanism. These findings provide further evidence that aggresome formation and autophagosome maturation both involve protein ubiquitination.
A Ubiquitin Code for Autophagy?
Ubiquitin chains on protein substrates are recognized and bound by specific ubiquitin-binding factors. Different ubiquitin chains can be created by forming linkage via different lysine residues in the ubiquitin molecule (lysine 6, 11, 27, 29, 33, and 48). Structural analyses have revealed that these ubiquitin chains adopt different conformations and, therefore in theory, would create distinct codes recognized by different ubiquitin-binding proteins. 38 Decades of studies have clearly established that proteins destined to proteasomes are polyubiquitinated predominantly through lysine 48 linkage. In yeast, it was shown that a proteasome-bound ubiquitinated protein is escorted by several Cdc48-dependent ubiquitin-binding complexes and finally handed to the ubiquitin receptors at the proteasomes. 39 Misfolded proteins are similarly tagged by K48-linked polyubiquitin chains for delivery to proteasomes. It can be inferred that misfolded proteins or aggregates are “redirected” to the aggresome and autophagy when they can no longer be processed by proteasomes, for example, upon treatment with a proteasome inhibitor. Do protein aggregates use a similar ubiquitin-dependent mechanism to enter the aggresome and become substrates for autophagy?
Using ubiquitin mutants that restrict ubiquitin chain formation to specific lysine residues, it was shown that p62 and HDAC6 both prefer binding to K63-linked ubiquitinated proteins. 18,40 This binding preference is supported by the quantitative analysis of ubiquitin-chain composition in the brain of p62 knockout mice, where a robust accumulation of K63-linked ubiquitin chains is apparent. 41 Similar accumulation of K63-linked ubiquitin chains is also found in HDAC6 knockout mouse embryo fibroblasts (MEFs) (unpublished observation). These results reveal that proteins targeted by p62 and HDAC6 are preferentially modified by K63-linked ubiquitin chains. Interestingly, the expression of a ubiquitin mutant that can only be conjugated via K63 leads to the stabilization of an artificial proteasome substrate, GFPu, without inhibiting proteasome activities. 42 This result suggests that K63-linked ubiquitination directs GFPu away from the proteasome. Indeed, forcing K63-linked ubiquitin chain formation also causes GFPu to accumulate to the aggresome. 42 These results suggest an interesting possibility that K63-linked ubiquitin chains, which are recognized by HDAC6 and p62, play an instructive role in rerouting the traffic of protein aggregates to the aggresome-autophagy machinery. K63-linked ubiquitin chains could be part of the defining ubiquitin code for QC autophagic substrates.
Surprisingly, the formation of aggresomes also requires a deubiquitinating enzyme ataxin 3, which can trim both K48- and K63-linked ubiquitin chains from proteins. 43,44 Interestingly, ataxin 3 also binds HDAC6. 43 In this context, ataxin 3 might “edit” ubiquitin chains on misfolded proteins, allowing the construction of new ubiquitin chains recognized by the associated HDAC6. Under this scenario, ubiquitin remodeling by a combination of trimming existing ubiquitin chains and adding new ones could create a new code that specifies protein trafficking to the aggresome and degradation by autophagy (Fig. 2).
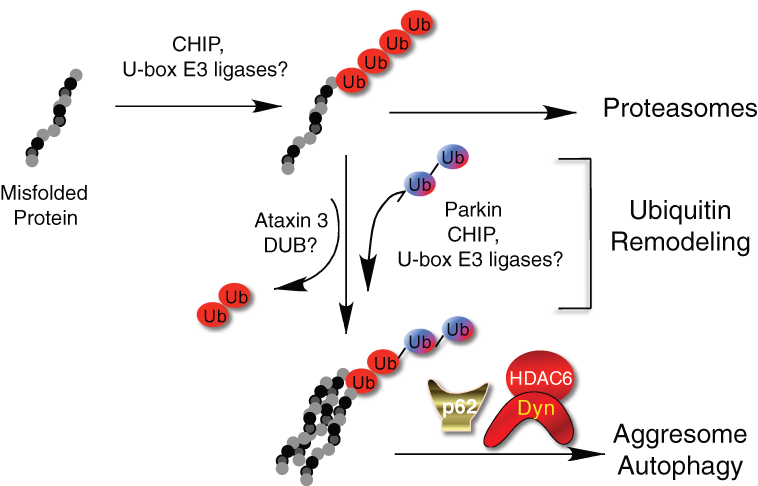
A model for the generation of autophagic/aggresomal ubiquitin codes. Misfolded proteins are ubiquitinated by chaperone-dependent E3 ligase CHIP or related E3 ligases. This ubiquitination, which is predominantly K48 linked, directs misfolded proteins to the proteasomes. Under certain stress conditions, the ubiquitin chains on misfolded proteins can undergo remodeling by the combined activity of deubiquitinating enzymes (DUB) and E3 ligases to remove and add ubiquitin chains of different linkages. The newly formed ubiquitin chains, which contain K63-linked chains, are recognized by p62 and HDAC6, which direct protein aggregates to the aggresome and autophagy. Dyn = dynein motor.
The Ubiquitin E3 Ligases in Autophagy-Dependent Protein Aggregate Processing
The ubiquitin code model predicts that the specific type of ubiquitin chains added to misfolded proteins determines to which degradative pathway they would be delivered. The ubiquitin E3 ligases responsible for the assembly of different ubiquitin chains on misfolded proteins are therefore keys to this process. Misfolded proteins are bound by molecular chaperones, such as Hsp70, to facilitate refolding. Interestingly, molecular chaperones also associate with specific ubiquitin E3 ligases. CHIP is the prototype of the chaperone-dependent ubiquitin E3 ligases. 45 CHIP binds both Hsp70 and Hsp90 and contains a U-box ubiquitin ligase domain. CHIP specifically catalyzes ubiquitination of denatured but not native proteins in a chaperone-dependent manner. 46 These findings support a simple model that misfolded proteins resistant to refolding by the molecular chaperone are ubiquitinated by CHIP-dependent K48-linked ubiquitination and degraded in proteasomes. 45 Interestingly, CHIP also binds UBC13, the ubiquitin E2 enzyme with K63 specificity. The CHIP-UBC13 complex can catalyze K63-linked ubiquitin chain formation, 47 suggesting a possibility that CHIP can also direct ubiquitinated proteins to the autophagic route. Indeed, it was reported that CHIP promotes degradation of α-synuclein, an aggregate-prone protein linked to Parkinson disease, by both proteasome- and lysosome-dependent mechanisms. 48 It is conceivable that CHIP and related E3 ligases might generate either the proteasomal or autophagic-targeting ubiquitin code by interacting with specific E2 conjugation enzymes or cofactors under different conditions (Fig. 2).
Biochemical and genetic studies show that CHIP also promotes the ubiquitination and degradation of the microtubule-associated protein, tau, whose aggregation leads to the formation of neurofibrillary tangles (NFTs), a pathological feature of various neurodegenerative diseases including Alzheimer disease. However, no apparent accumulation of tau aggregates was detected in CHIP knockout mice. 49 This finding could reflect functional redundancy of other E3 ligases involved in protein aggregate clearance. Several U-box–type ubiquitin E3 ligases related to CHIP have been identified. Interestingly, similar to CHIP, these U-box E3 ligases all associate with molecular chaperones or cochaperones. 50 Because of their association with molecular chaperones, CHIP and related U-box–containing E3 ligases might play an important role in the elimination of misfolded proteins or aggregates. The potential involvement of other U-box E3 ligases in protein aggregate clearance remains to be investigated.
Ubiquitin and Other Forms of QC Autophagy
Parkin is another K63-linked E3 ligase that has been intensely studied for its role in misfolded protein processing. Mutations in parkin are the most common causes of early-onset familial Parkinson disease (AR-JP). 51 There are reported AR-JP cases without Lewy body pathology, suggesting a potential link of parkin to the formation of Lewy bodies and protein aggregate clearance. 52 Interestingly, parkin was reported to associate with CHIP to form a more active E3 ligase complex. 53 It is possible that these two E3 ligases work in conjunction to control protein aggregate clearance. Despite a large body of literature on this subject, a definitive proof that parkin plays a dominant role in protein aggregate clearance by autophagy remains elusive.
However, convincing evidence has emerged to support a critical function of parkin in the clearance of impaired mitochondria by autophagy. 54 This so-called mitophagy requires parkin to ubiquitinate “mitochondria.” 55,56 AR-JP–causing parkin mutants, including those deficient in E3 ligase activity, are defective in supporting mitophagy. These findings indicate that inability to clear damaged mitochondria could be a critical basis of Parkinsonism caused by parkin mutations. This proposition is entirely consistent with the prominent mitochondrial defect observed in Parkinson disease patients and parkin-deficient animal models. 57
Similar to protein aggregate clearance, parkin-mediated ubiquitination of mitochondria recruits both p62 and HDAC6. Disease-causing E3 ligase–deficient parkin mutants that cannot promote mitochondrial ubiquitination also fail to concentrate p62 and HDAC6 to depolarized mitochondria. 56 In this case, parkin-mediated ubiquitination is clearly required for the clearance of mitochondria by autophagy. 55,56 The identification of a parkin-equivalent E3 ligase in protein aggregate clearance would be essential to unambiguously establish a role of ubiquitin modification in protein aggregate clearance.
During mitophagy, juxtanuclear mitochondrial aggregates that resemble protein aggregate–induced aggresomes are formed. 55,56,58 The formation of these “mito-aggresome” structures requires HDAC6 as well as microtubule motor-dependent transport. That the same ubiquitin-binding p62 and HDAC6 as well as cortactin-dependent actin remodeling are required for the clearance of protein aggregates and damaged mitochondria suggests a common mechanism in the execution of QC autophagy. 56 Whether a universal ubiquitin code recognized by HDAC6 and p62 is responsible for all forms of selective QC autophagy, including ribophagy (for ribosomes) and xenophagy (for bacteria), is a critical question whose answer would have important implications in human health.
Aggresome Formation and QC Autophagy
Aggresome formation is likely part of the integral response to excessive production of protein aggregates or damaged mitochondria, a situation that occurs during the progression of neurodegenerative disease. In both cases, disruption of the microtubule network and aggresome formation suppresses the clearance of these two toxic entities. 29,56,59 Therefore, the concentration of these toxic entities to the MTOC region facilitates their clearance. Consistent with this idea, autophagic vesicles and lysosomes are found concentrated around the aggresome. 29,60 Accordingly, the transport of protein aggregates to the MTOC might facilitate their access to lysosomes and autophagosomes. 60 The concentration of lysosomes to the MTOC region also requires HDAC6. 60 It is clear that the transport of protein aggregates and autophagic components is highly coordinated to ensure productive autophagy. Therefore, the formation of the aggresome is an integral part of QC autophagy central to efficient disposal of protein aggregates.
mTOR and Autophagy-Dependent Protein Aggregate Clearance
HDAC6 is not required for the induction of autophagosomes induced by misfolded protein stress. 29 Evidence also indicates that p62 is not required for autophagosome formation induced by mitochondrial stresses. 61 How is autophagy activated to clear protein aggregates? mTOR is a central protein kinase that regulates cellular growth in response to nutrient availability and intracellular energy status. Logically, active mTOR inhibits autophagy when nutrients are abundant. 62 Although mTOR-independent autophagy pathways clearly exist, 63 characterization of the mTOR signaling has played an instrumental role in establishing the relevance and utility of autophagy in the disposal of protein aggregates. 64 The mTOR inhibitor rapamycin, an immunosuppressant widely used in clinics, is a potent activator of autophagy. In several neurodegenerative animal models, rapamycin treatment suppresses protein aggregate accumulation and neurodegeneration, supporting a critical role of mTOR-regulated autophagy in protein aggregate clearance. 6,64 Indeed, misfolded protein stress and agents that induce QC autophagy all inhibit mTOR, specifically, mTORC1 activity. 65-67 Accordingly, misfolded protein or other QC autophagy stresses would induce autophagosomes by inhibiting mTOR activity. The autophagosomes are then recruited to ubiquitinated substrates by p62 and further processed by HDAC6. Supporting this idea, in the Drosophila SBMA model, the neuroprotective effect of rapamycin was abrogated in the HDAC6 mutant background. 6 Thus, induction, targeting, and maturation of autophagosomes are independently regulated. Given that HDAC6 is not required for starvation-induced autophagy, 29 this finding would also suggest that rapamycin activates QC and not starvation-induced autophagy. Although the signaling mechanism that distinguishes QC and starvation autophagy remains unknown, the mTOR kinase complexes would likely play an instructive role.
The Clinical Implication of Autophagy-Dependent Protein Aggregate Clearance
Neurodegenerative Disease
The finding that neural-specific ablation of atg5 or atg7, two genes essential for autophagy, leads to accumulation of ubiquitin-positive protein aggregates and progressive loss of neurons in mice has provided definitive proof that autophagy is required for the clearance of protein aggregates and protects individuals from neurodegeneration. 68,69 If protein aggregate toxicity is the key basis of neuronal dysfunction or death in neurodegenerative disease, enhancing autophagy to eliminate protein aggregates would be a logical therapeutic approach. The antineurodegenerative activity of rapamycin provides a proof of principle to this concept. However, the potent immunosuppressive activity of rapamycin is a major barrier for its clinical utility in neurodegenerative disease patients. Not surprisingly, “autophagy activators” are hotly pursued as potential therapeutic agents for neurodegenerative diseases. 70 However, as illustrated in HDAC6-deficient cells and Drosophila, activation of autophagy or formation of autophagosomes does not necessarily lead to productive autophagy. 6,29 Accordingly, if neurodegenerative patients have defects in the maturation of autophagosomes or recognition of protein aggregates, 71 an induction of autophagy might have only marginal effects in overall protein aggregate clearance. The abundant accumulation of autophagosomes in neurons affected by neurodegenerative diseases could, in fact, reflect normal induction but defective maturation of autophagosomes. In a detailed morphological analysis, Nixon et al. reported an abnormal accumulation of immature autophagosomes in the brain from Alzheimer disease patients. 72 It was speculated that the fusion of autophagosomes to lysosomes was impaired in the affected individual. Similar autophagosome-lysosome fusion defects might also be linked to a familial form of frontal-temporal dementia (FTD) caused by mutations in CHMP2B. CHMP2B is a component of multivesicular body (MVB), which sorts and delivers membrane proteins to lysosomes via a ubiquitin-dependent mechanism. Overexpression of the FTD-associated CHMP2B mutant or siRNA-mediated knockdown of key MVB components, such as TSG101, suppresses autophagosome-lysosome fusion and causes ubiquitin-positive protein aggregate accumulation. 73 As discussed previously, neurodegenerative IBMPFD can also be traced to a defect in autophagosome-lysosome fusion caused by p97/VCP mutations. Determining the prevalence of autophagosome-lysosome fusion defects in neurodegenerative patients could have potential impacts on developing an effective therapy. For example, combining an autophagy activator with a fusion promoter could be a more effective strategy to eliminate protein aggregates and provide therapeutic relief to neurodegenerative patients.
Cancer
Given their robust protein synthesis and high mutation rate, tumor cells are expected to produce higher amounts of misfolded proteins than their normal counterparts. This is particularly true for tumor cells with high secretory capacity, such as multiple myeloma, which synthesize and secrete immunoglobulin that are subject to extensive posttranslational modification and therefore prone to errors and misfold. It is possible that pharmacological disruption of protein quality control machinery would lead to excessive accumulation of toxic protein aggregates, leading to tumor cell death. The remarkable clinical efficacy of a proteasome inhibitor, bortezomib, on the otherwise incurable multiple myeloma might be a result of a dramatic accumulation of misfolded immunoglobulin in tumor cells. Using a proteasome substrate reporter, it was shown that an increase in autophagy activity can restore proteasome activity that was inhibited by aggregation-prone protein. 6 This result clearly demonstrates that the proteasome and autophagy are complementary degradative machinery that disposes of misfolded proteins and aggregates. A combination of a proteasome inhibitor with an aggresome or autophagy inhibitor would in theory block the two complementary pathways, resulting in excess protein aggregate accumulation. Supporting this view, inhibition of aggresome formation by the HDAC6-selective inhibitor tubacin enhanced the killing effect of bortezomib on multiple myeloma cell lines. 74 It is expected that combined inhibition of proteasome and autophagy machinery would also have more potent antitumor effects. The elucidation of how misfolded protein aggregates are processed has provided a novel therapeutic opportunity for cancer.
Concluding Remarks
It is widely assumed that ubiquitin associated with protein aggregates are the leftover marks on the misfolded proteins originally tagged for proteasomes. The demonstration of critical roles for ubiquitin-binding proteins, p62 and HDAC6, in aggresome formation and protein aggregate clearance provides the crucial clue to the discovery that ubiquitin modification is, in fact, central to autophagy-dependent protein aggregate clearance. While the details of the aggresomal and autophagic ubiquitin code remain to be elucidated, it is apparent that a coordinated action of ubiquitin E3 ligases, deubiquitinating enzymes, and ubiquitin-binding factors is responsible for directing protein aggregates to their final clearance station. These findings not only further illustrate the remarkable regulatory power of ubiquitin-dependent modification but also reveal an unexpected complexity of autophagy. Given the nature of QC autophagy in maintaining healthy proteins and organelles, this machinery would likely play a major role in human health and disease. The ability to modulate QC autophagy could hold promise to improve therapies targeting many forms of age-related diseases.
Footnotes
Acknowledgements
The author thanks Dr. K.L. Lim and Ms. A.W. McClure for comments and suggestions.
This work was supported by the National Institutes of Health (NS053825).
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.