
Abstract
Conjugation of ubiquitin (ubiquitination) to substrate proteins is a widespread modification that ensures fidelity of many cellular processes. During mitosis, different dynamic morphological transitions have to be coordinated in a temporal and spatial manner to allow for precise partitioning of the genetic material into two daughter cells, and ubiquitination of key mitotic factors is believed to provide both directionality and fidelity to this process. While directionality can be achieved by a proteolytic type of ubiquitination signal, the fidelity is often determined by various types of ubiquitin conjugation that does not target substrates for proteolysis by the proteasome. An additional level of complexity is provided by various ubiquitin-interacting proteins that act downstream of the ubiquitinated substrate and can serve as “decoders” for the ubiquitin signal. They may, specifically reverse ubiquitin attachment (deubiquitinating enzymes, DUBs) or, act as a receptor for transfer of the ubiquitinated substrate toward downstream signaling components and/or subcellular compartments (ubiquitin-binding proteins, UBPs). In this review, we aim at summarizing the knowledge and emerging concepts about the role of ubiquitin decoders, DUBs, and UBPs that contribute to faithful regulation of mitotic division.
Conjugating and “Decoding” Ubiquitin
Conjugation of ubiquitin is one of the major posttranslational modifications of proteins in eukaryotic cell. Highly dynamic and reversible, ubiquitination modulates and orchestrates a broad range of cellular processes, including protein degradation, quality control and trafficking, signal transduction, differentiation, and cell division.1-4 In this pathway, ubiquitin is covalently attached to a substrate by coordinated cycles of 3 enzymatic reactions, ubiquitin activation (E1 enzyme), ubiquitin conjugation (E2 enzyme), and ubiquitin ligation (E3 ubiquitin ligase) (Fig. 1). One of the mostly described consequences of ubiquitination is degradation of the substrate proteins by a large protease complex, 26S proteasome. Due to its irreversible character, this process provides essential directionality toward various pathways. Importantly, ubiquitination of the targeted substrates does not always serve as a signal for the proteasomal degradation but may also regulate protein localization, binding to other proteins, or even enzymatic activities. 5 The covalent linkage of single ubiquitins (mono- ubiquitination or multi-ubiquitination) to the substrates or various modes of ubiquitin binding to the already attached ubiquitin moiety (polyubiquitin chain formation) determine the fate of the substrate protein (Fig. 1). A huge variation of the polyubiquitin chains can be formed, as one of the 7 internal lysine residues or the N-terminal methionine can serve as attachment sites in a single ubiquitin molecule. This creates a plethora of possible ubiquitin signals.
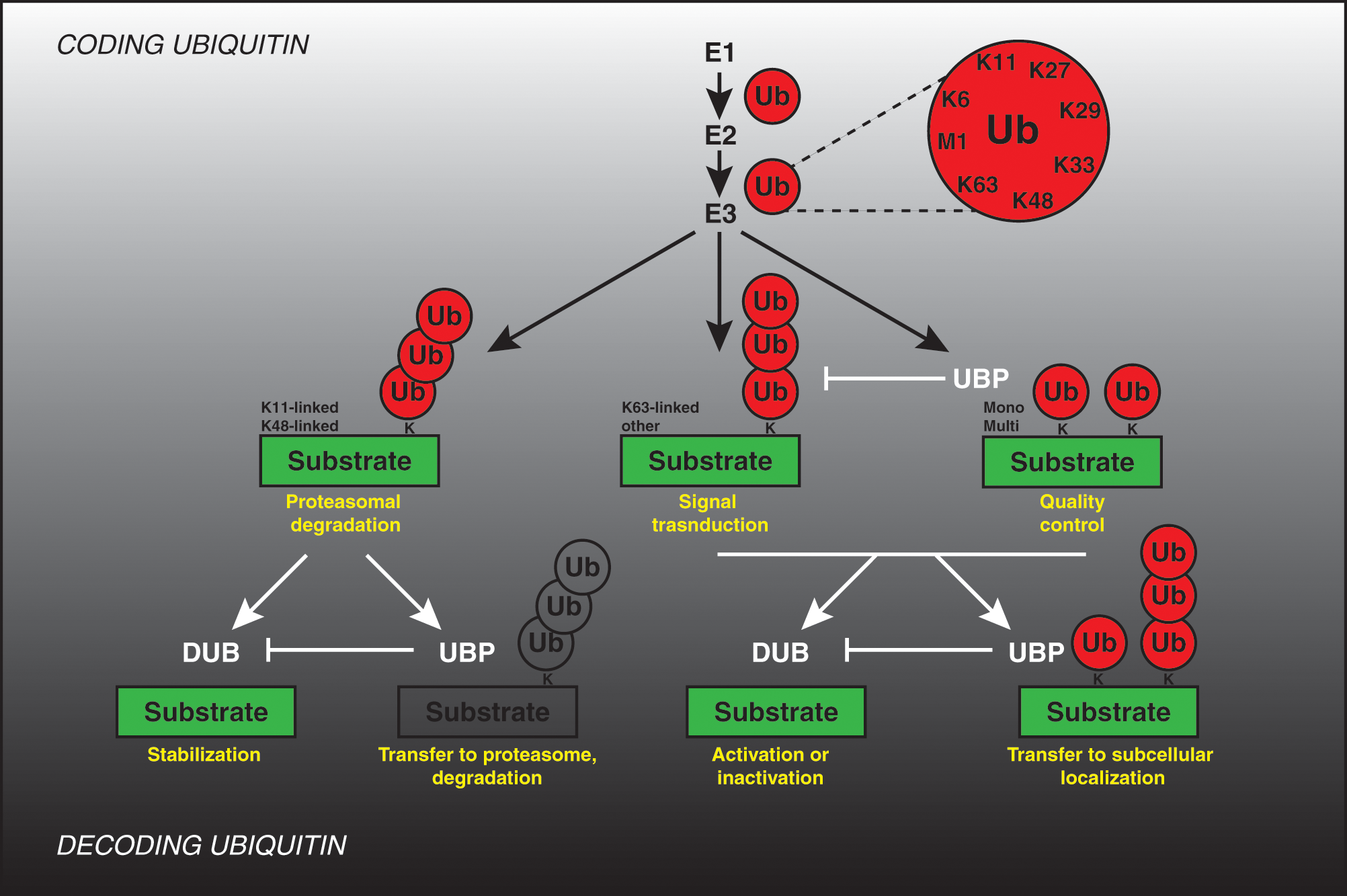
Coding and decoding ubiquitin. Coding: Ubiquitin (Ub) is covalently attached to the lysine residues (K) of substrate proteins by a 3-step mechanism involving the sequential actions of E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) enzymes, resulting in formation of both mono- and multi-ubiquitination signal as well as formation of the poly-ubiquitin chains linked via one of the internal lysine residues or N-terminal methionine residue. The possible physiological outcomes of different ubiquitination signals are indicated in yellow. Decoding: Ubiquitin modifications may be removed by specific de-ubiquitinating enzymes (DUB) and may stabilize substrates or lead to their activation or inactivation (yellow). Ubiquitin-binding proteins (UBP) interact with ubiquitinated proteins and may prevent the conversion of mono-ubiquitin into polyubiquitin chains, protect ubiquitin modifications from DUBs, target proteins to the 26S proteasome, and/or mediate downstream signaling events perhaps through new protein–protein interactions or targeting them to subcellular compartments (yellow).
An additional level of complexity is provided by various ubiquitin-interacting proteins that act downstream of the ubiquitinated substrate and can serve as “decoders” for the ubiquitin signal. They may, specifically reverse ubiquitin attachment (deubiquitinating enzymes, DUBs) or, act as a receptor for transfer of the ubiquitinated substrate toward downstream signaling components and/or subcellular compartments (ubiquitin-binding proteins, UBPs) (Fig. 1). Eukaryotic genomes encode for high numbers of both ubiquitin conjugation machinery and “decoders” of ubiquitin signal, constituting a network which by analogy to the signal transduction pathways should ensure a high complexity regulation of cellular processes. It is predicted that human cells express about 600 different E3-ubiquitin ligases, 100 DUBs, and as many as 200 various UPBs. Yet, the precise roles and substrate specificity of these factors are only beginning to be understood. In this review, we aim at summarizing the recently emerging concepts and knowledge about the role of ubiquitin decoders during regulation of mitosis. Excellent existing reviews have described the roles of the E3-ubiquitin ligases known to act during mitotic division,6-8 and likewise the role of ubiquitin coding and decoding machinery in regulation of other cell cycle stages has been extensively described.9-13 Thanks to the high-throughput, whole genome screening efforts in mammalian cells and genetic studies in yeast, we have started to unravel novel mitotic factors that decode versatile ubiquitin signal.
Mitosis
During the mammalian cell cycle, genetic material has to be duplicated and then undergo mitosis, in which the two copies of each chromosome are segregated into two daughter cells (Fig. 2). Each of the daughter cells must receive an exact copy of the genetic material, as defects in chromosome segregation can cause genetic instability and aneuploidy, which has been linked to tumorigenesis. 14 Thus, for successful mitosis, a precise coordination of the morphological changes with time has to be accomplished. Onset of mitosis is typically marked by the nuclear envelope breakdown and condensation of the replicated DNA during prophase. The individualized chromosomes assemble their kinetochores during prometaphase and metaphase stages. The assembly of the mitotic spindle and its attachment to the sister kinetochores allows proper chromosome segregation. Accurate chromosome segregation requires that sister kinetochores attach to microtubules from opposite spindle poles (bipolar attachment) in metaphase. Kinetochore attachment is a stochastic process and as such is prone to errors, which may result in chromosome misalignment. A complex network of regulatory factors constituting so-called spindle assembly checkpoint (SAC) ensures that sister chromatid segregation in anaphase does not take place before all chromosomes are properly aligned at the metaphase plate and before their kinetochores are under sufficient occupancy and tension by spindle microtubules.15-18 Following successful chromosome segregation, the spindle microtubules undergo a dramatic reorganization, and the anaphase central spindle or spindle midzone is formed. During this transition, the antiparallel, interdigitating microtubules and many associated proteins become organized into discrete bundles in the spindle midzone. Midzone microtubules play an important role in cytokinesis, which separates the cytoplasm of the two daughter cells.19-21 Cytokinesis begins with the assembly of a contractile actin-myosin ring. Its contraction results in the formation of the midbody, which is composed of the remnants of the spindle midzone. In the final step, the actomyosin ring disassembles and the plasma membranes resolve in a process called abscission. 22 An accepted view is that cytokinesis cannot be completed if chromosomes are pulled apart erroneously or if the anaphase spindle midzone is not formed properly. 23 In budding yeast, these defects lead to the activation of the recently described “NoCut” checkpoint pathway. 24 In mammalian cells, abscission also fails in such cases, leading to regression of the cleavage furrow and the formation of multinucleated cells.19-21
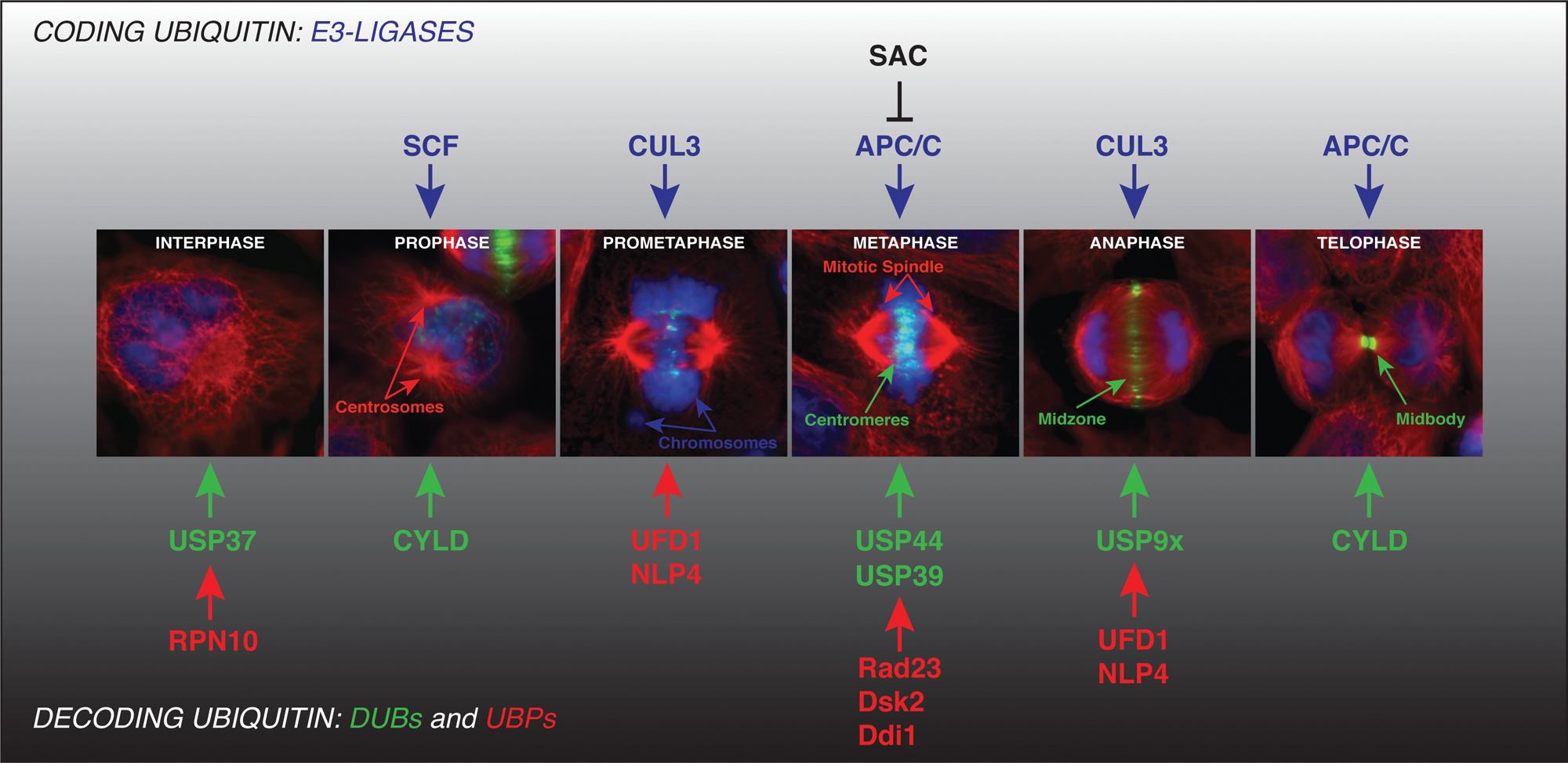
Regulation of mitosis by ubiquitin system. Faithful regulation of mammalian mitotic division (prophase, prometaphase, metaphase, anaphase, and telophase) requires precise coordination of structural transitions. This machinery involves many critical factors and enzymes that need to be precisely regulated in time and space. Thus, both their timely expression and correct localization to the subcellular compartments must be achieved during different morphological transitions. Blue indicates nucleus and chromosomes; green (representing the CPC component, Aurora B) marks the centromere structures during prometa- and metaphases and midzone and midbody regions during anaphase and telophase, respectively; and red marks microtubules and spindle structures. The critical and major ubiquitin E3-ligases (blue) responsible for coding ubiquitin on mitotic factors are depicted. APC/C E3-ligase is a subject of fidelity control by the spindle assembly checkpoint (SAC) network. The ubiquitin decoders, DUBs (green) and UBPs (red), contribute to fidelity and directionality of mitotic division at specific stages and transitions (for details, see text).
Two major E3-ligases are known to control cell division: SCF-complexes regulate the G1 to S-phase transition, and the anaphase-promoting complex/cyclosome (APC/C) coordinates metaphase to anaphase transition and mitotic exit. However, recent findings hint at a more complex picture of regulation of this process, and accumulating evidence suggests a role of SCF ligases in controlling the early mitotic phases. 25 Likewise some novel cullin-based E3s, like CUL3, essentially contribute to regulation of mitosis26,27 (Fig. 2).
Role of DUBs During Mitotic Progression
The human genome encodes for about 100 different DUBs, which belong to 5 distinct families. Four of them—USPs (ubiquitin-specific proteases), UCHs (ubiquitin C-terminal hydrolases), OTUs (ovarian tumor), and Josephin family—are cysteine proteases, whereas the fifth DUB family comprises a group of Zn2+ metalloproteases, which are referred to as JAB1/MPN/MOV34 (or JAMMs).28-30 In addition to DUBs acting mostly on substrates modified by ubiquitin, specific enzymes exist that act on other ubiquitin-like molecules (UBLs), but their roles are poorly understood thus far. 31 DUBs have been implicated in many physiological and pathophysiological processes, including apoptosis, DNA repair, neuro degenerative diseases, and cancer, but their precise substrates and regulation are known only in some cases. Here, we summarize the DUBs with known functions during the cell division cycle (Table 1) and use examples of DUBs required specifically for mitosis to illustrate complexity of their regulation.
List of DUBs Regulating Cell Cycle
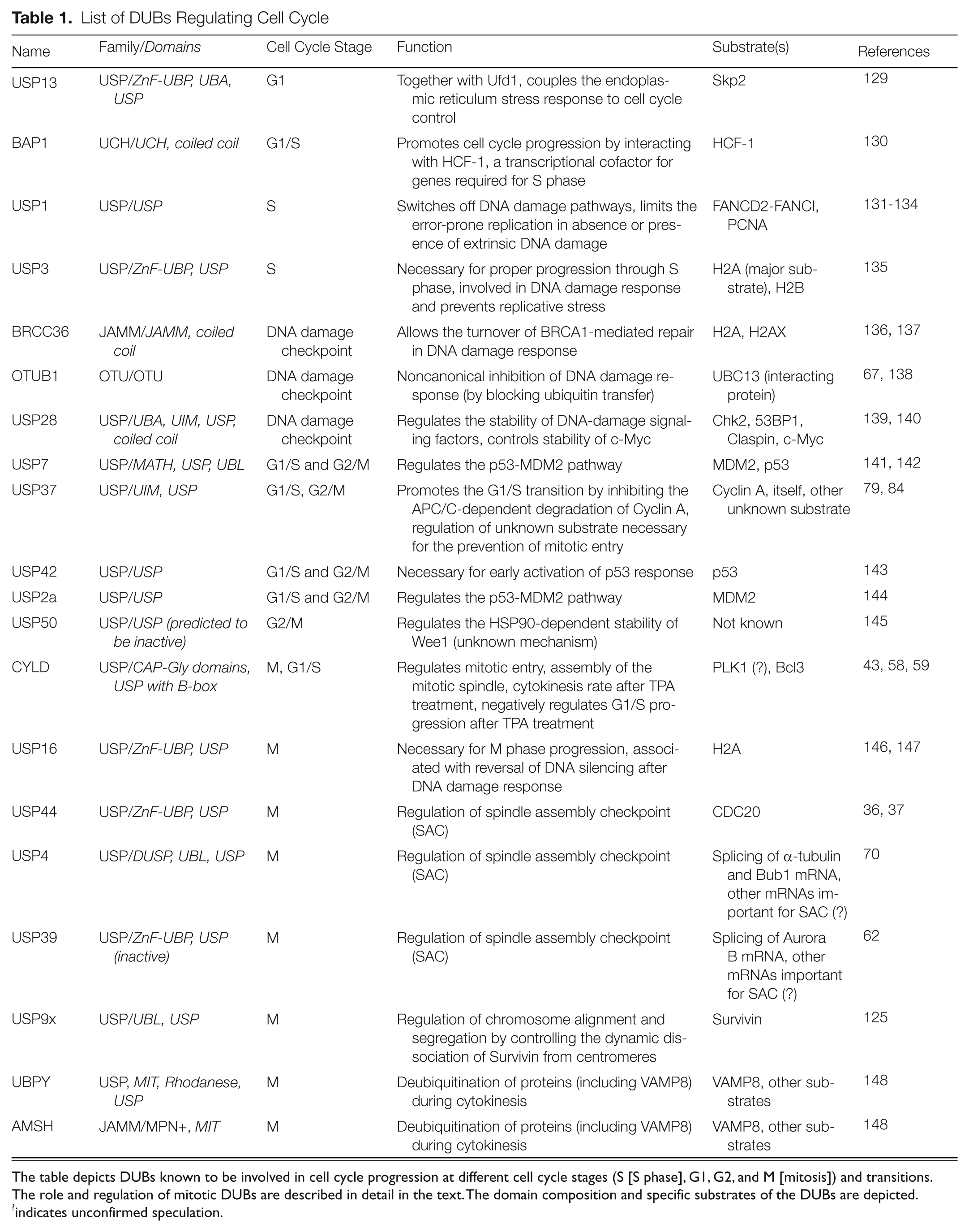
The table depicts DUBs known to be involved in cell cycle progression at different cell cycle stages (S [S phase], G1, G2, and M [mitosis]) and transitions. The role and regulation of mitotic DUBs are described in detail in the text. The domain composition and specific substrates of the DUBs are depicted.
indicates unconfirmed speculation.
Recent mass spectrometry experiments suggest that as many as 5,000 of ubiquitinated substrate proteins may exist in human cells, with 19,000 possible modification sites. 32 Thus, evolutionary pressure could exist that allowed for expansion of the DUB family, ensuring a high level of specificity of these enzymes. Indeed, inactivation of many DUBs leads to very specific phenotypes in cells and organisms, which could arise from the ability of these enzymes to recognize and bind specifically to the diverse ubiquitination signals (Fig. 1).
DUBs Reversing Nonproteolytic Ubiquitination Signal
USP44
The SAC ensures that mitosis does not go beyond metaphase until all chromosomal kinetochores are correctly attached to spindle microtubules, allowing for faithful chromosome segregation and partition of genetic information between the two daughter cells. Both occupancy of kinetochores by the spindle microtubules and tension generated by bipolar attachment between sister kinetochores are sensed by the SAC. 18 Activated SAC inhibits the APC/C E3-ubiquitin ligase by sequestering its major mitotic co-activator, CDC20 protein.33,34 This is achieved by formation of the mitotic checkpoint complex (MCC) composed of the main effector SAC protein, MAD2, along with BubR1, Bub3, and CDC20. 35 Binding of MAD2 to CDC20 occurs at unattached kinetochores, but many other regulatory checkpoint components are required to ensure and strengthen the inhibitory effect of the SAC. Attachment of all kinetochores releases SAC inhibition, allowing APC/CCDC20 to target Securin (inhibitor of sister chromatid separation) and Cyclin B proteins for proteolytic ubiquitination, leading to the anaphase onset and mitotic exit. Stegmeier et al., 36 aiming to identify novel components of SAC in human cells, screened a shRNA library targeting approximately 800 genes related to ubiquitin-signaling for their ability to bypass the spindle checkpoint arrest induced by microtubule poison paclitaxel. One of the identified hits was the DUB, USP44. Indeed, co-transfection of the USP44 siRNA and the siRNA-resistant construct of wild-type USP44 (but not catalytically inactive enzyme) results in rescue of the spindle checkpoint defect. Moreover, inactivation of USP44 in untreated cells leads to premature anaphase onset and chromosomal segregation defects, suggesting that deubiquitinating activity of USP44 is required for efficient SAC signaling and anaphase timing. Intriguingly, SAC defect observed in USP44-depleted cells was not caused by a defect in kinetochore recruitment of checkpoint components. Instead, binding of MAD2 to APC/CCDC20 was perturbed in these cells, suggesting that USP44 maintains the mitotic arrest by stabilizing the association between MAD2 and CDC20. Interestingly, in another parallel study, APC/C-dependent, nonproteolytic ubiquitination of CDC20 has been shown to be responsible for the disassembly of the MAD2-CDC20 complexes. 37 This CDC20 ubiquitination event is mediated by the UbcH10 E2-conjugating enzyme and is required for activation of the APC/C. Consistent with these findings, Stegmeier et al. have shown that in mitotic extracts, USP44 inhibits the ubiquitination activity of APC/C. Consequently, inactivation of UbcH10 in USP44-depleted cells leads to reduction of ubiquitinated CDC20, restoration of MAD2-CDC20 association, and rescue of the mitotic checkpoint arrest. These results suggest that USP44 acts as an antagonist of UbcH10 toward APC/C’s own-dependent ubiquitination of CDC20. Thus, the SAC is regulated through a dynamic balance of APC/C-dependent nonproteolytic ubiquitination and USP44-dependent deubiquitination. During final stages of metaphase, when not all chromosomal kinetochores are occupied by spindle microtubules, the SAC is activated through a USP44-dependent “safeguard” mechanism, which by deubiquitinating CDC20 stabilizes MAD2-CDC20 complexes. Thus, the SAC is built on two antagonistic pathways, which can switch rapidly from one to the other, sensing the occupancy state of microtubules on kinetochores.
How is this fine balance between ubiquitination and deubiquitination of CDC20 regulated? It has been shown that USP44 protein levels peak in mitotic cells and are decreased immediately after chromosome attachment to mitotic spindles. 36 Moreover, USP44 is also phosphorylated in early mitosis and immediately dephosphorylated after SAC satisfaction but before degradation of Cyclin B. The RNA polymerase II carboxy terminal domain phosphatase, Fcp1, has been shown to mediate dephosphorylation of USP44. 38 Fcp1 is a transcription regulator, and it has been shown to be an antagonist to CDK1 in Aspergillus nidulans, 39 where expression of a defective Fcp1 allele together with an inhibitory phosphorylation-resistant CDK1 allele induced severe mitotic defects including impaired nuclear separation. Mitotic extracts have been used to show that the phosphorylation status of USP44 modulates its activity: Dephosphorylation by Fcp1 leads to the reduction of USP44 activity, which is associated with a dramatic decrease of the MAD2-CDC20 association and mitotic exit. Thus, the phosphorylation status of USP44 may govern the dynamic balance between ubiquitination and deubiquitination of CDC20.
The model by Stegmeier et al. 36 of the mitotic checkpoint regulation by antagonistic ubiquitination and deubiquitination has been challenged by another study. 40 According to this work, in checkpoint-activated HeLa cells, CDC20 is ubiquitinated by APC/C, leading to its proteasomal degradation. Indeed, ubiquitination-dependent degradation of yeast Cdc20 homolog during activated checkpoint has been demonstrated in Saccharomyces cerevisae. 41 Moreover, a CDC20 lysine-less mutant allows the inhibition of the mitotic checkpoint by promoting the mitotic exit, which is more consistent with the model in which ubiquitination-dependent degradation of CDC20 maintains the SAC. 40 This mitotic exit does not happen immediately, as it has been shown in checkpoint-arrested cells expressing the CDC20 lysine-less mutant, as these cells have an active SAC during at least 2 hours, suggesting that additional ubiquitin-independent mechanism is required to sustain the checkpoint arrest. Reddy et al. 37 have also observed a partial proteolysis of CDC20 upon addition of UbcH10 and ubiquitin on checkpoint-arrested extracts; however, this proteolysis event does not regulate stability of the MCC complex. The other point is that the CDC20 lysine-less mutant binds more strongly to the checkpoint proteins MAD2 and BubR1 than the wild-type CDC20 in checkpoint-arrested cells, suggesting that CDC20 ubiquitination could modulate the binding to MAD2 and BubR1 as suggested by the model of Stegmeier et al. However, Nilsson et al. 40 argued that this strong association between the CDC20 lysine-less variant, MAD2, and BubR1 is due to an increased affinity upon the exchange of a conserved lysine into the arginine residue in the MAD2 binding site of CDC20. In principle, two mechanisms of maintaining the SAC signaling are not mutually exclusive and could work at the same time: CDC20 associated with the MCC components could be ubiquitinated by APC/C, allowing its dissociation from the MCC, and it would be degraded subsequently by the proteasome. In such a scenario, CDC20 should be progressively modified by two different types of ubiquitin signal: first a nonproteolytic and then proteolytic one. The action of USP44 would be required to “proofread” ubiquitination status of CDC20 and prevent assembly of inappropriate ubiquitin chains. Thus, future studies need to clarify the type of ubiquitin signals present on CDC20 during the precise time periods of SAC response. Interestingly, overexpression of USP44 in mouse embryonic cells has been shown to increase the association between MAD2 and CDC20 and to lead to an anaphase delay consistent with role of USP44 in the regulation of SAC. 42
CYLD
The same shRNA-based screen that led to identification of USP44 revealed the role of another DUB, CYLD, as a regulator of early mitotic phases. 43 Mutations in the tumor suppressor gene CYLD cause genetic predisposition to human familial cylindromatosis, 44 and CYLD deubiquitinating enzyme was shown to negatively regulate NFκB, JNK,45-47 and Wnt signaling pathways. 48 Some in vitro studies have suggested that CYLD is able to cleave specifically K63-linked and linear ubiquitin chains. 49 In the study by Stegmeier et al., 43 CYLD knockdown led to accumulation of nonmitotic cells after paclitaxel treatment. These cells had a normal interphase nuclear morphology rather reflecting a delay in the mitotic entry process. This phenotype was independent of the regulatory function of CYLD in the NFκB pathway. The premitotic arrest phenotype is rescued by the expression of wild-type CYLD but not an inactive CYLD mutant, suggesting that deubiquitinating activity of CYLD is required for its early mitotic function. Consistently, CYLD-depleted cells are characterized by delayed accumulation of Serine 10-phosphorylated histone H3, delay in phosphorylation of CDC25C, and delayed degradation of Emi1. Intriguingly, downregulation of the mitotic kinase, Polo-like kinase 1 (PLK1), led to the same phenotypes and delayed both CDC25C phosphorylation and mitotic entry.50-52 Interestingly, PLK1 has been identified as a binding protein of CYLD using proteomic approaches, suggesting a cooperative role of both proteins in the early mitotic pathway. To give a functional explanation for the delayed entry into mitosis in CYLD-downregulated cells, the authors argued that CYLD could be involved in the “prophase” checkpoint, which has been proposed to regulate entry into early mitotic phases 53 and which delays chromosome condensation in response to impaired microtubule structure. The key component of this pathway is CHFR (checkpoint with FHA and RING domains), which is an E3-ubiquitin ligase.54-56 It has been shown that in vitro, CHFR is able to generate K63-linked ubiquitin chains in cooperation with the E2 conjugating heterodimer Ubc13-MMS. 56 Thus, it is tempting to propose that CYLD antagonizes the CHFR activity in order to restart the progression of the cell cycle after resolving microtubule defects. Indeed, based on experiments with Xenopus egg extracts, it has been proposed that PLK1 is ubiquitinated by CHFR and then degraded in order to establish a delay in the entry of mitosis upon mitotic stress. 54 Whether CHFR ubiquitinates PLK1 in cells and which kind of ubiquitin signal is used are not known. Alternatively, other, unknown components of the prophase checkpoint signaling modified by CHFR-mediated K63-linked ubiquitination could be direct potential substrates of CYLD. Thus, further studies are required to confirm the involvement of CYLD in the prophase checkpoint.
Interestingly, it has been shown that stable expression of CYLD in U2OS cells results in an increase of fragmented nuclei and multinucleated cells, reflecting impairment in chromosome segregation and/or cytokinesis. 43 Furthermore, localization analysis has shown that overexpressed CYLD is associated with microtubules in interphase and at the midbody in telophase stage, suggesting another potential mitotic function of CYLD. Consistent with these observations, in addition to its USP domain, CYLD contains 3 cytoskeletal-associated protein-glycin-conserved (CAP-Gly) repeats. CAP-Gly domain is a conserved motif found in a large number of microtubule-associated proteins and could explain microtubule localization of CYLD. Indeed, CYLD interacts with the microtubule subunit tubulin through its two first CAP-Gly independent of its deubiquitinating activity.57,58 This interaction could allow regulation of the microtubules’ dynamics by CYLD, as CYLD depletion in HeLa cells induces a slow microtubule regrowth after nocodazole treatment, 57 and its overexpression leads to accelerated microtubule regrowth in melanoma cells. 58 Furthermore, the midbody localization of CYLD has been confirmed at the level of endogenous protein in primary mouse keratinocytes. 58 Because many proteins involved in the regulation of cytokinesis are localized to the midbody, CYLD could have a potential function in this process. It has been shown that CYLD co-localizes at the midbody with the histone deacetylase HDAC6, leading to its inhibition and high levels of acetylated, more stable microtubules, suggesting that CYLD could participate in the regulation of microtubule dynamics during the final stages of cytokinesis. However, since CYLD’s role in regulation of acetylation status of microtubules does not involve its deubiquitinating activity, it is not clear how exactly CYLD exerts its function at the midbody at the mechanistic level.
However, CYLD seems also to play a role in the assembly of the mitotic spindle. 59 This function could be linked to the interaction of CYLD with the centrosomal protein CEP192, which is involved in the centrosome maturation and nucleation of microtubules.60,61 In CEP192-depleted cells, microtubules are assembled in the vicinity of chromosomes and are unable to self-organize into bipolar spindles. Co-depletion of CEP192 and CYLD alleviates the spindle assembly defects, suggesting that CYLD function could be inhibited by CEP192. Because CYLD regulates microtubule dynamics in interphase cells, Gomez-Ferreria et al. 59 proposed that the potential inhibition of CYLD by CEP192 could take place at the onset of mitosis to allow for depolymerization of microtubules and/or to maintain possible K63-linked ubiquitination-mediated interaction(s) necessary for the spindle assembly.
Taken together, CYLD appears to fulfill some important functions during both early mitotic stages and mitotic exit. Further studies are needed to identify the specific and direct targets of this DUB as well as the precise modes of its regulation during mitosis.
USP39 and USP4
Using a RNAi-based screen, van Leuken et al. 62 identified USP39 as another regulator of the SAC. Knockdown of USP39 in synchronized U2OS cells leads to an increase in the cellular DNA content reflecting a defect in chromosome segregation and/or cytokinesis. Interestingly, the USP39-depleted cells were able to bypass the mitotic arrest induced by treatment with a microtubule stabilizing drug, paclitaxel, but not by nocodazole, which induces microtubule depolymerization. As mentioned previously, SAC is activated not only by the presence of unattached kinetochores but also by a lack of tension exerted across sister centromeres. Indeed, paclitaxel decreases the interkinetochore tension, suggesting that USP39 is specifically required to sustain the tension-dependent branch of the SAC. USP39 is also known as a 65 kDa SR-related protein of the U4/U6.U5 tri-snRNP. It associates with small ribonuclear proteins and is involved in the splicing process. 63 van Leuken et al. 62 found that depletion of USP39 leads to a decrease of the mRNA levels of the mitotic kinase Aurora B and consequently its protein levels. Ectopic transfection of Aurora B cDNA restored Aurora B protein levels in USP39-depleted cells but failed to rescue the mitotic arrest upon paclitaxel treatment. These observations suggested that USP39 is a critical regulator of the tension-dependent mitotic checkpoint through its function in the splicing of the Aurora B mRNA and possibly other mRNAs of essential mitotic factors. Aurora B kinase is a catalytic component of the so-called chromosomal passenger complex (CPC), containing INCENP, Survivin, Borealin/Dasra B, and Telophase-Disc-60 (TD60) proteins. Aurora B is able to correct erroneous microtubule-kinetochore attachments in particular monotelic and syntelic attachments where not enough tension is generated between sister chromatids.64-66
Although USP39 contains an ubiquitin-protease domain, the cysteine and histidine residues belonging to the catalytic triad of the DUB’s cysteine-protease are not conserved, and consequently the authors have reported absence of the catalytic activity of USP39 in vitro. Despite the fact that USP39 might be catalytically inactive, it could participate in an important molecular pathway. Indeed, other studies have postulated that some DUBs do exert a function in a noncatalytic manner. For example, OTUB1 inhibits DNA double-strand breaks independently of its catalytic activity, 67 and yeast Ubp6 is involved in regulation of the proteasome-dependent degradation in a noncatalytic manner. 68 These studies highlight the possibility that DUBs that have been predicted to be inactive, such as USP50 and USP52 to 54, 69 exert noncanonical DUB functions, possibly by acting as ubiquitin-binding receptors rather than processing enzymes.
Another elegant study demonstrated the role of splicing in SAC signaling and identified DUB USP4 as a regulator of the spliceosome function during SAC response induced by treatments with paclitaxel and monastrol (a specific inhibitor of kinesin Eg5 required for formation of bipolar spindles). 70 Downregulation of USP4 leads to SAC bypass after these treatments, and mRNA levels of α-tubulin (constituent of microtubules) and Bub1 (SAC protein) are reduced in these cells. The assembly of spliceosome is initiated by binding of the U1 snRNP to the 5′ splice site followed by recognition of the branch point site and 3′ splice site by SF1/BBP and U2AF, respectively. 71 The prespliceosome is formed after recruitment of the U2 snRNP. The U4/U6 and U5 snRNPs are then added as preassembled U4/U6.U5 tri-snRNP to form an inactive complex. Upon release of U1 and U4 snRNPs, splicing reactions start. After the mRNA splicing, the spliceosome is disassembled, allowing another round of splicing. USP4 together with its substrate targeting factor, the U4/U6 recycling factor Sart3, is able to deubiquitinate the Prp3 protein, 70 which is a component of the U4 snRNP. Prp3 ubiquitination is catalyzed by the U- box-containing protein Prp19, which is a member of the Prp19 complex (Nine Teen Complex, or NTC). In yeast, this complex participates in the splicing process by influencing the biogenesis of U4/U6 snRNP and stabilizing the binding of U5 and U6 snRNPs on the spliceosome after U4 release.72,73 The ubiquitination signal of the Prp3 protein is nonproteolytic, and these K63-linked ubiquitin chains are then recognized by the U5 snRNP component, Prp8. Indeed, it has been previously shown that yeast Prp8 is able to bind ubiquitin in vitro through its variant Jab1/MPN domain. 74 This interaction is necessary for the stabilization of the U4/U6.U5 tri-snRNP, confirming previous study about the requirement of ubiquitin for U4/U6.U5 stability. 75 The working model suggests that Prp3 ubiquitination acts on U4/U6.U5 stabilization during the formation of inactive spliceosomal complex. After recruitment of the U4/U6.U5 tri-snRNP to the spliceosome, USP4-Sart3 deubiquitinates Prp3, facilitating the release of the U4 snRNP necessary for activation of the splicing machinery. To highlight the requirement of splicing for the fidelity of cell division, Song et al. 70 showed that depletion of several other spliceosomal factors leads to mitotic defects in HeLa cells treated with paclitaxel drug. Interestingly, knockdown of the 65 kDa SR-related protein of the U4/U6.U5 tri-snRNP, USP39, is responsible for the SAC bypass, confirming results obtained by van Leuken et al. 62 Thus, USP39 and USP4 regulate splicing and are indirect but critical regulators of the mitosis.
Altogether, these studies suggest that splicing is a very dynamic process and that the spliceosome is a subject of constant and profound conformational rearrangements. Throughout the splicing cycle, distinct RNAs and proteins are assembled and disassembled at precise time points. Thus, modifications of proteins by ubiquitination allow for modulation of these highly dynamic rearrangements in the spliceosome composition. Both reversibility of ubiquitin modification and binding of the specific ubiquitin receptor proteins to the nonproteolytic ubiquitin chains allow for regulation of stable protein complexes and subcomplexes during splicing. These studies also highlight the possibility that other dynamic cellular processes could be regulated by ubiquitination of the splicing machinery.
Interestingly, the usp4 gene belongs to a chromosomal region that is frequently deleted in small-cell lung cancer (SCLC). Furthermore, SCLC is associated with reduced expression of USP4 76 and alterations of ploidy. 77 Because defects in the SAC often lead to increase in polyploidization, loss of USP4 could be linked to tumorigenesis.
DUB Reversing Proteolytic Ubiquitination Signal
USP37
Irreversible degradation by the ubiquitin-proteasome system confers directionality of the cell cycle. The APC/C E3 ligase together with its co-activators (CDC20 and CDH1) orchestrates cell cycle progression through mitosis and G1 phases 6 (Fig. 2). The CDC20 activates APC/C during mitotic phases, whereas CDH1 activates APC/C as cells exit mitosis and in G1 phase. This sequential activation manner, also referred to as “substrate ordering,” allows the temporal order of degradation of APC/C substrates such as Cyclin A, Securin, Cyclin B, Geminin, PLK1, and Aurora A 78 and ensures faithful cell cycle progression. By counterbalancing the activities of E3 ubiquitin ligases, DUBs could be involved in the timing of degradation of the ubiquitination substrates. To identify potential DUBs implicated in the regulation of substrates of APC/C, Huang et al. 79 used a proteomic approach. Interestingly, USP37 has been found to interact specifically with the co-activator CDH1 but not with CDC20. 79 Furthermore, APC/C subunits such as CDC27, APC5, and APC7 co-immunoprecipitated with the endogenous USP37, and interaction with CDC27 is decreased upon CDH1 knockdown, suggesting that USP37 binds to APC/C through the CDH1 co-activator. Interestingly, overexpression of USP37 in synchronized U2OS cells led to an accumulation of Cyclin A protein level, which could reflect a reduced rate of its proteasomal degradation. Moreover, USP37-overexpressing cells are also characterized by an accelerated entry into S phase. Conversely, cells expressing specific USP37 shRNAs exhibited a delay in accumulation of Cyclin A after release from nocodazole-induced mitotic block and were also delayed for the entry into S phase. Thus, USP37 may regulate turnover of Cyclin A and thereby G1/S transition. Furthermore, Huang et al. 79 have also demonstrated that endogenous USP37 is able to bind to Cyclin A, suggesting that Cyclin A could be a direct USP37 substrate. Indeed, unlike catalytically inactive mutant, the wild-type form of USP37 is able to reduce ubiquitination of Cyclin A in cells and to counterbalance the activity of APC/CCDH1 in vitro. 79 These results strongly establish USP37 as an antagonist of APC/CCDH1 toward ubiquitination of Cyclin A, allowing for rescue from its proteasomal degradation and faithful progression beyond the G1/S transition. However, how is the function of USP37 regulated to ensure temporal specificity? By analyzing the USP37 protein levels, the authors observed that USP37 fluctuates in a cell cycle–dependent manner. USP37 levels peaked in the late G1 phase, decreased in late mitosis, and reappeared in G1 again, reflecting a typical profile of degradation events governed by APC/CCDH1 at the M/G1 transition. It is known that APC/CCDH1 recognizes substrates via short D- or KEN-box motives.80,81 Indeed, USP37 contains a KEN-box motif necessary for the K11-linked ubiquitination mediated by APC/CCDH1 and its subsequent degradation. Interestingly, USP37 is also able to rescue itself from degradation in late G1. Thus, USP37 acts as an APC/CCDH1 substrate in late mitosis, and it counterbalances the APC/CCDH1 activity toward itself and Cyclin A in late G1. These opposing functions are modulated by CDK2-mediated phosphorylation of USP37. In their working model, the authors present evidence that Cyclin E-CDK2 phosphorylates USP37 in late G1, resulting in its activation and its rescue from degradation. USP37 acts also on ubiquitinated Cyclin A, which in turn associates with CDK2 and amplifies the activation of USP37. This positive feedback loop promotes the G1/S transition. The USP37 activation is reinforced by phosphorylation-dependent inhibition of APC/CCDH1 by Cyclin A-CDK2. 82 In late mitosis, cyclin partners were degraded, preventing phosphorylation-mediated activation of USP37. At the same time, activated APC/CCDH1 is responsible of the proteolytic ubiquitination of USP37. The work by Huang et al. nicely illustrates the dynamic interplay between a DUB and E3-ubiquitin ligase, which is necessary for timely regulation of the key cell cycle regulators. In a recent proteomic survey it was found that indeed many DUBs associate with E3-ligase complexes, 83 suggesting that cross-regulation observed between USP37 and APC/CCDH1 could be a commonly used mechanism for spatiotemporal regulation of other important biological pathways. It will be interesting to understand whether additional DUBs exist that specifically counteract ubiquitination of some of the other numerous targets of APC/C. Likewise, the role of putative novel DUBs that may act on substrates of the SCF E3-ligases during early mitotic phases, and targets of the recently described CUL3-based E3-ligases coordinating mitotic progression, are currently unknown.
Recently, Burrows et al. 84 demonstrated that USP37 is also subjected to a second wave of degradation during the cell cycle. Indeed, a fraction of USP37 is degraded by the SCFβTrCP E3 ligase at the G2/M transition. This degradation event requires a PLK1-dependent phosphorylation at a noncanonical degron within the sequence of USP37. Interestingly, the expression of a SCFβTrCP-resistant USP37 mutant leads to a delay of the mitotic entry, suggesting that one of the USP37 pools regulates unknown substrates required for the inhibition of entry into mitosis. This USP37 pool does not seem to regulate Cyclin A. 84 Thus, identification of the new USP37 substrate will be essential to understand the cell cycle functions of both USP37 pools.
UBPs in Mitosis
Recognition of ubiquitinated substrates is mediated by a variety of ubiquitin-binding proteins (UBPs) that serve as ubiquitin receptors (or decoders) and contain at least 1 ubiquitin-binding domain (UBD) within their structure.85-87 Based on their structural features, UBDs can be divided into several subgroups, such as ubiquitin-associated (UBA) domain, ubiquitin-interacting motif (UIM), motif interacting with ubiquitin (MIU), and many others. 88 The common feature of these UBDs is the ability to noncovalently bind ubiquitin moiety. Increasing experimental evidence suggests that UBPs play a critical role in defining the fate of ubiquitinated targets in time and space, either by mediating their proteasomal degradation or by regulating their localization and/or interaction with other proteins. UBPs have recently been also shown to play a role in mitosis (Fig. 2). Rad23, Dsk2, Ddi1, and Rpn10 were identified in Saccharomyces cerevisiae as regulators of mitotic progression by shuttling ubiquitinated targets to the proteasome for degradation,89-93 whereas the second group includes UBPs mediating relocalization of its targets involving the Cdc48/p97-Ufd1-Npl4 (p97) complex in mammalian cells. Although the p97 complex was proposed to play a role in chromosome segregation and spindle dynamics,94-96 very little is known about specific UBPs controlling mitosis in mammalian cells, in particular the functional homologs of Rad23 pathway. Here, we summarize the role of UBPs controlling cell cycle (Table 2), with the main focus on known yeast UBPs and their unpredicted redundancy in controlling mitotic progression. Furthermore, we discuss the role of human p97 complex during mitosis.
List of UBPs Regulating Cell Cycle
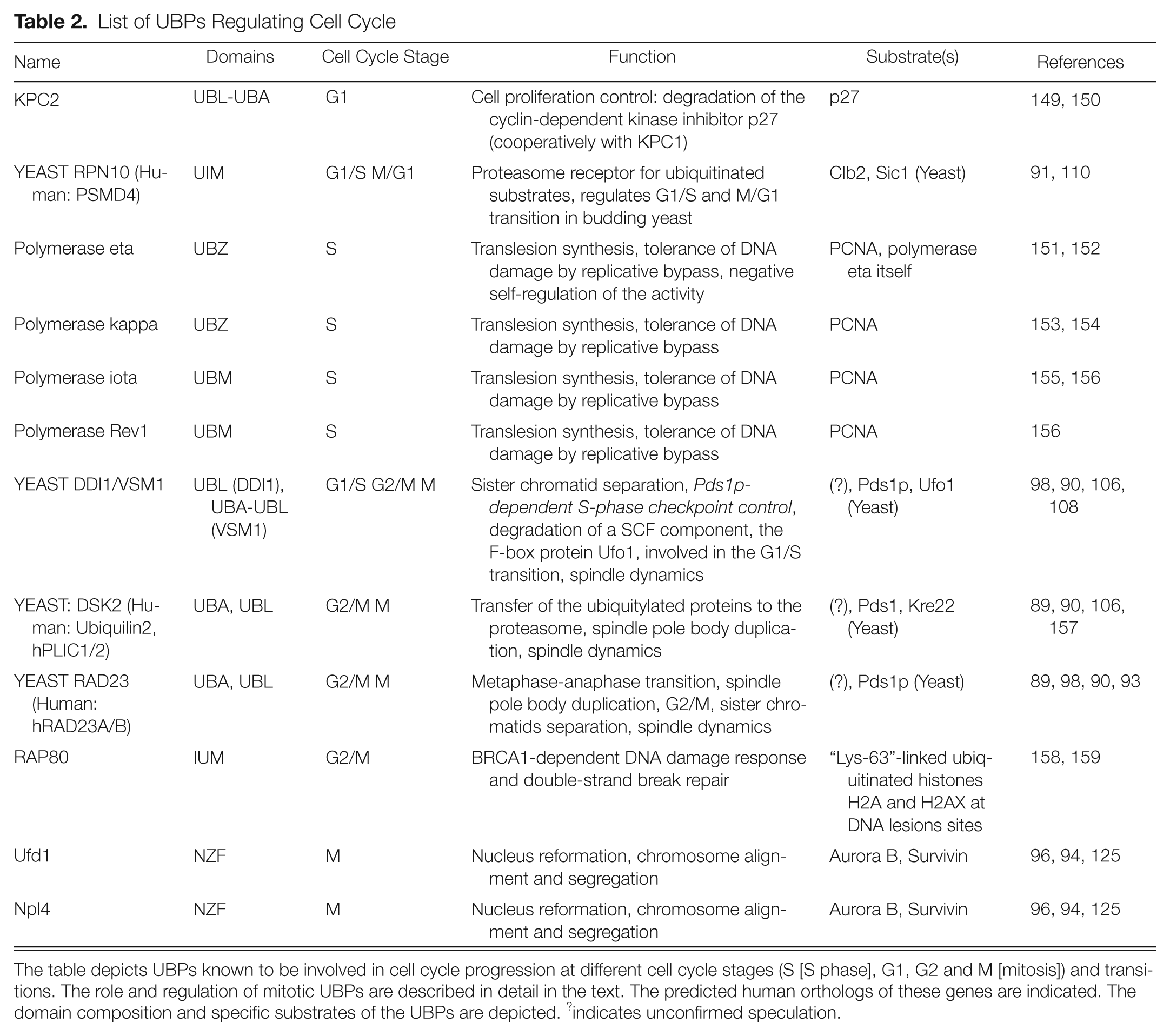
The table depicts UBPs known to be involved in cell cycle progression at different cell cycle stages (S [S phase], G1, G2 and M [mitosis]) and transitions. The role and regulation of mitotic UBPs are described in detail in the text. The predicted human orthologs of these genes are indicated. The domain composition and specific substrates of the UBPs are depicted. ?indicates unconfirmed speculation.
UBPs Targeting Substrates for Proteasomal Degradation
Rad23, Dsk2, and Ddi1
In yeast Saccharomyces cerevisiae, Rad23 and Ddi1 were shown to shuttle ubiquitinated proteins to the 26S proteasome for degradation. 93 These proteins belong to UBPs that possess an UBA domain (2 UBA domains in case of Rad23) interacting with ubiquitin and ubiquitinated substrates.97,98 In addition, Rad23 and Ddi1 have the N-terminal ubiquitin-like domain (UBL) that is able to interact with the proteasome.99,100 RAD23 and DDI1 genes were initially identified in a screen for suppressors of the temperature sensitive mutant allele of PDS1, pds1-128. 90 Pds1 (securin in higher vertebrates) is an essential regulator of mitosis and inhibitor of anaphase onset.101,102 Proteasomal degradation of Pds1 releases its inhibitory action on Separin, Esp1 (Separase in higher vertebrates) that mediates cleavage of cohesin complex, sister chromatid separation, and anaphase spindle elongation, ensuring successful completion of mitosis in yeast cells. 103 Pds1 is polyubiquitinated by APC/C prior to the onset of anaphase, which leads to recognition of the ubiquitin chain by 26S proteasome and degradation of Pds1 at the beginning of anaphase. 104 Interestingly, deletions of both RAD23 and DDI1 genes were able to rescue pds1-128 temperature sensitive phenotype, suggesting a possible role of these proteins in the regulation of Pds1 proteolysis during mitosis. 90 Furthermore, the mechanism of anaphase control was proposed in which Rad23 and Ddi1 bind ubiquitinated Pds1, thus inhibiting ubiquitin chain elongation and subsequent Pds1 proteolysis. 90 It is not known however, what is the mechanism of Rad23-Ddi1-Pds1 complex dissociation that finally leads to Pds1 degradation by proteasome.
The third UBA-UBL protein, Dsk2, was also shown to play a role in governing ubiquitinated substrates to proteasome.99,105 Overexpression of Dsk2 is toxic for cells due to accumulation of ubiquitinated substrates and leads to mitotic arrest, an abnormal nuclear positioning, and affected spindle dynamics.89,99 There are controversial data about involvement of Dsk2 in spindle pole body (centrosomes in mammalian cells) duplication. Some studies have proposed a model according to which Dsk2 together with Rad23 assists the assembly of Cdc31 into the spindle pole body, which is considered to be an essential event in its duplication. 89 However, a follow-up study has shown that deletion of Rad23 and/or Dsk2, as well as triple deletion of Rad23-Ddi1-Dsk2, did not lead to the spindle pole body duplication failure. 106 Interestingly, it has been shown that Rad23, Ddi1, and Dsk2 exert partially redundant roles in cell cycle progression. 106 Double deletions of Rad23-Dsk2 and Rad23-Ddi1 showed slight accumulation of cells in G2/M; however, single deletions of these 3 genes and also double deletion of Ddi1-Dsk2 did not show any defect in mitotic progression. Furthermore, triple deletion Rad23-Ddi1-Dsk2 led to striking cell cycle arrest either in G2 or in anaphase. Additionally, spindle dynamics were shown to be affected in triple deletion strain; however, the underlying mechanisms still remain unclear. 106 According to these results, Rad23 shares a common function with Ddi1 and Dsk2 in mitotic control, but it is not clear how these proteins are regulated to coordinate the common tasks. One possible level of such regulation that could allow for specificity in binding of the ubiquitinated substrates is formation of both homo- and heterodimers between Rad23, Ddi1, and Dsk2 proteins.105,107 UBA and/or UBL domains were shown to be essential in Rad23 and Dsk2 homodimerization and Rad23 heterodimerization with Dsk2 and Ddi1.93,107,108 According to one of the proposed models, dimerization of UBA/UBL proteins can potentially play a role in preventing unnecessary ubiquitin chain elongation or premature disassembly during the transit of substrates to the proteasome: Polyubiquitin chain of a substrate might be tightly covered with a complex containing multiple UBA domains, thus blocking access of E3-ligases and/or deubiquitinated enzymes to the substrate. 93 However, the biological significance of UBPs dimerization still remains to be determined experimentally.
In addition to the role in mitosis, Ddi1 was shown to be implicated in the negative regulation of the late secretory pathway by interacting with exocytic v- and t-SNAREs. 108 It is not yet clear how Ddi1 coordinates both processes, in particular, which stimuli can possibly serve as a switch between these two functions by regulating the change in localization of Ddi1 in the cell (either on a plasma membrane or in the nucleus).
Taken together, these results suggest a role of UBA-UBL proteins Rad23, Ddi1, and Dsk2 in controlling mitotic progression. These proteins were shown to inhibit degradation of ubiquitinated Pds1, thus delaying the onset of anaphase, and to influence spindle dynamics. The regulatory functions of UBPs are complicated by redundancy and cooperation between ubiquitin receptors. Thus, the exact mechanisms of mitotic control by Rad23, Ddi1 and Dsk2 remain to be elucidated. Future studies are needed to identify other yet unknown targets of Rad23-Ddi1-Dsk2 proteins that play a role during mitotic progression. Likewise, the functional orthologs of this pathway in higher vertebrates need to be characterized.
Rpn10
The Rad23-pathway components are not the only UBPs known to play a role in the regulation of mitosis in yeast. Another UBP, Rpn10, was shown to play a role in determining the fate of critical mitotic factors. Rpn10 is a stoichiometric component of 26S proteasome, and it was predicted to be one of the ubiquitin receptors governing ubiquitinated proteins for degradation.109,110 Rpn10 contains 2 domains: N-terminal von Willebrand A domain, which is necessary for interaction with proteasome, and C-terminal ubiquitin-interacting motif (UIM) domain, which ensures Rpn10 binding to the ubiquitinated targets. Among other targets, Rpn10 controls turnover and degradation of one of the key cell cycle regulators, Sic1.91,110 Sic1 is a specific stoichiometric inhibitor of Cdk1/Clb (CDK/Cyclin in mammalian cells) complexes that are required for S-, G2-, and M-progression and promote spindle pole body separation in yeast.111,112 Sic1 is synthesized at the end of mitosis and degraded at the beginning of S-phase in a Rpn10-dependent manner.110,113 During late stages of mitosis, Sic1 inhibits Cdk1/Clb2 activity and is considered to be an activator of M/G1 transition, although the precise mechanisms need to be elucidated.114-117 At the same time, there is some evidence that other ubiquitin receptors, in particular Rad23-Ddi1-Dsk2 proteins (and other UBPs), can share the same function of shuttling targets for degradation with Rpn10, as deletion of Rpn10 in yeast is not lethal and recombinant Rad23 is able to rescue Rpn10 deletion phenotype to some extent. In contrast, deletion of Rpn10 in Drosophila melanogaster results in pupal lethality, and siRNA-mediated knockdown of Rpn10 in Trypanosoma brucei leads to G2/M cell cycle arrest.91,118,119 Thus, the redundancy with other UBPs and the precise role of different domains of Rpn10 remain the unresolved issues that deserve extensive study.
UBPs Regulating Substrates Localization
p97 complex
Cdc48/p97-Ufd1-Npl4 is the evolutionally conserved protein complex that plays a role in the so-called ERAD (endoplasmic reticulum-associated degradation) pathway. It binds ubiquitinated proteins and is necessary for the export of the misfolded proteins from the ER to the cytoplasm, where they are degraded by the proteasome. 120 In this complex, the AAA+ ATPase Cdc48/p97 (also known as VCP in vertebrates) interacts with its adapters Ufd1-Npl4, which are able to bind ubiquitin through their NZF (Npl4 Zn-F) domains.121-123 Studies performed in Xenopus laevis egg extracts revealed that apart from its function in the ER, the p97 complex regulates spindle disassembly at the end of mitosis and is necessary for formation of a closed nuclear envelope. This occurs through removal of ubiquitinated mitotic kinase Aurora B, a component of the CPC complex, from chromatin by Ufd1-Npl4 adapters during the late stages of mitosis. 96 In contrast to this observation, in HeLa cells Ufd1-Npl4 adapters of the p97 complex were shown to antagonize chromosome-associated Aurora B activity already during earlier mitotic stages, which resulted in defects in faithful chromosome segregation as cells progressed through mitotic division. 94 These observations are consistent with the model in which Aurora B and possibly other components of CPC are ubiquitinated on the mitotic chromosomes and “extracted” by the action of p97 complex. It is not clear, however, what is the fate of the ubiquitinated Aurora B bound to p97 complex or how this association is regulated. Aurora B kinase is indeed a subject of APC/C-mediated, proteolytic ubiquitination, but this event takes place during later stages of mitotic exit. 124 Interestingly, Aurora B was shown to be ubiquitinated earlier during mitosis by Cul3-based E3-ligase, but in contrast to the APC/C-mediated modification, this ubiquitination event does not seem to influence protein stability of Aurora B but rather its faithful relocalization to mitotic spindle.26,27 It is not clear at this point whether Ufd1-Npl4 adapters localize to the mitotic spindle of whether they may change the affinity of the ubiquitinated CPC toward microtubules. It is necessary to mention that results of another study performed in HeLa cells suggested that Ufd1 rather recruits Aurora B to chromosomes during mitosis. 125 Additionally, the authors have identified a putative DUB, USP9x, involved in regulation of dynamics of another CPC component, Survivin protein. Using siRNA, immunostaining, and FRAP techniques, investigators showed that USP9x regulates dissociation of Survivin from the centromeres and thereby chromosome alignment, segregation, and completion of cytokinesis. 125 However, the protein levels of Survivin were not affected by downregulation of USP9x. Interestingly, it has been shown that USP9x specifically hydrolyzes both Lys 29 and Lys 33 linked polyubiquitin chains, 126 which leads to regulation of activity of AMPK-related kinases and not their proteolysis. It will be interesting to uncover the precise subcellular localization of USP9x in cells during mitotic progression.
Despite evident difficulty in reconciling these observations, it is possible that different CPC components are regulated by several cooperating pathways, involving modification by different ubiquitination signals, proofreading by USP9x deubiquitinating enzyme, and finally dissociation from chromosomes by the action of p97 complex or yet to be identified microtubule-associated UBPs. Intriguingly, several other studies performed in worm Caenorhabditis elegans did not show the requirement of p97 complex for mitotic progression.127,128 Overall, Cdc48/p97-Ufd1-Npl4 complex is considered to play a role in mitosis in vertebrates, but the exact function of ubiquitin-binding adapters Ufd1-Npl4 in this process requires further studies.
Concluding Remarks and Future Perspectives
Recent progress in study of the ubiquitin pathways has uncovered an enormous versatility and flexibility of this signaling network. This complexity is reflected not only by a plethora of different ubiquitination signals that can be attached to the substrates but also by the fact that their conjugation may take place at a very precise time points during the physiological processes and in a very specific subcellular localizations. This image is further complicated by so-called “decoders” of the ubiquitin signal, which may, on one hand, reverse ubiquitination like DUBs or, on the other hand, transfer the modified substrate to the downstream pathways as in the case of UBPs. For both large family of proteins, at least two functional groups can be distinguished: one that regulates proteasomal degradation (rescue or transfer, respectively) and another that can process the nonproteolytic type of ubiquitination signals and regulates substrate localization, activity, and/or stability of protein complexes (Fig. 1). Despite the recent advances in understanding the components of this network in regulation of mitosis, the current list of DUBs and UBPs involved in mitotic progression is far from complete. Thus, identification of new key players in this field will significantly improve our understanding of the principles of mitotic regulation. Likewise, major efforts are needed to understand the precise modes of regulation of these new “decoders,” which could guarantee the essential specificity toward the substrates. We believe that development of imaging technologies, in particular specific “ubiquitin-fluorescent sensors” that could allow visualization of attachment of specific ubiquitin signals in a cellular context, will greatly advance our knowledge about this fascinating signaling network.
Footnotes
Acknowledgements
The authors wish to thank the members of the entire Sumara group for careful reading of the manuscript and for helpful discussions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research in the laboratory of Izabela Sumara is sponsored by IGBMC, ATIP-AVENIR program from CNRS and INSERM, Program de Mecenat from Sanofi-Aventis, Foundation Research Medical (FRM), Region Alsace, and University of Strasbourg.