
Abstract
The cullin family of ubiquitin ligases can potentially assemble hundreds of RING-type E3 complexes (CRLs) by utilizing different substrate receptors that share common interaction domains. Cullin receptors dictate substrate specificity, and cullin-mediated substrate degradation controls a wide range of cellular processes, including proliferation, differentiation, and apoptosis. Dysregulation of cullin activity has been shown to contribute to oncogenesis through the accumulation of oncoproteins or the excessive degradation of tumor suppressors. In this review, we will discuss cullin complexes and their substrates, the regulatory pathways that affect cullin activity, and the mechanisms by which cullins may facilitate or inhibit carcinogenesis.
Introduction
Ubiquitin-mediated proteolysis of cellular proteins plays a vital role in maintaining the balance between normal growth and uncontrolled proliferation. Precisely controlled protein ubiquitination and degradation are required for orderly progression through the cell cycle and to respond to intrinsic and extrinsic signals. The cullin family of ubiquitin ligases, comprised of CUL1, 2, 3, 4A, 4B, 5, and 7, represents the largest class of RING-type E3 ligases (CRLs) through the diverse array of substrate receptors that assemble with and impart specificity to the modular CRL complexes. Cullins do not have intrinsic catalytic activity but instead serve as scaffolds that facilitate the assembly of multimeric E3 ligase complexes and transfer ubiquitin from the E2-conjugating enzyme to the substrate. The RING finger protein Rbx1/ROC1/Hrt1 mediates the interaction between the cullin C-terminus and the E2 enzyme, while substrate binding occurs at the N-terminus. An adaptor protein may be utilized to mediate the interaction between the cullin N-terminus and the substrate receptors that impart specificity to the cullin complexes.
Additional posttranslational modifications of substrates, such as phosphorylation or hydroxylation, may be required for recognition by certain substrate receptors as a means of regulating protein ubiquitination (reviewed in Petroski and Deshaies 1 ). Moreover, all cullin-based ubiquitin ligases themselves are regulated by Nedd8 (a ubiquitin-like protein) posttranslational modification, which is carried out in a manner analogous to ubiquitination (reviewed in Pan et al. 2 ). Nedd8 modification is required for cullin activation to induce a conformational change that increases the proximity between the E2 enzyme and the substrate 3 and prevents the inhibitory association of the cullin-associated neddylation-dissociated protein (CAND1). 4 The COP9 signalosome is a multimeric complex that serves to deneddylate activated cullins, 5 thus restoring CAND1-mediated inhibition of the ubiquitin ligase complexes. Additional regulators have also been found for specific cullins. For example, CUL4-based E3 complexes enlist accessory factors to promote substrate ubiquitination, such as DET1 for c-Jun and the nonreceptor tyrosine kinase c-Abl for DDB2 ubiquitination. 6,7 Cullins themselves are also ubiquitinated, and deubiquitinating enzymes (e.g., Ubp12) have been described that counteract the destabilizing effect of cullin ubiquitination. 8
Cullins are not conventional onco-proteins or tumor suppressors, as their effect on oncogenesis is dictated by their substrate receptors and corresponding substrates. There are multiple indirect mechanisms by which cullins contribute to carcinogenesis. Mutations in substrate receptors or substrates that result in the accumulation of oncoproteins drive proliferation and confer survival and genomic instability. Dysregulation of signaling pathways can affect substrate degradation through additional posttranslational modifications that alter substrate recognition by its receptor. Tumor-causing viruses (e.g., human papillomaviruses) promote transformation by hijacking the cellular ubiquitination machinery, including cullin-based E3 ligases, to direct the degradation of tumor suppressors. Therefore, cullin-based ubiquitin ligases orchestrate a wide array of cellular regulatory mechanisms, and malfunctions or perturbations of the intricate balance of cellular pathways may contribute to the multistep process of malignant transformation and tumorigenesis.
CUL1
To date, CUL1 is the most extensively characterized member of the cullin family. The core CUL1 ubiquitin ligase complex (denoted SCF) contains Rbx1, which binds the Cdc34 E2-conjugating enzyme, and Skp1, which acts as an adaptor for the F-box–containing substrate receptors (reviewed in Petroski and Deshaies 1 ). In Caenorhabditis elegans, knockdown of CUL1 by siRNA yielded hyperplasia of all tissues. 9 Knockout of Cul1 in mice resulted in early embryonic lethality, likely from the pleiotropic effects of CUL1 substrate accumulation. 10,11 Therefore, CUL1 plays an essential role in embryonic development and contributes significantly to normal and malignant cellular processes.
CUL1-Skp2
The extensively studied F-box protein Skp2 assembles an SCF complex with Skp1 and CUL1 to target the cell cycle inhibitor p27/Kip1 for degradation. 12 Skp2 amplification and overexpression have been observed in a wide array of cancers and are correlated with poor prognosis (reviewed in Hershko 13 ). Knockout of Skp2 in mice led to markedly enlarged nuclei with polyploidy and multiple centrosomes, indicating a critical role of Skp2 in controlling the abundance of cell cycle regulators. 14 The overreplication phenotype of Skp2 knockout mice is rescued by breeding with p27–/– mice, 15 indicating that p27 is the predominant substrate of SCFSkp2. Additional cell cycle regulators targeted by the SCFSkp2 ubiquitin ligase include the cyclin-dependent kinase inhibitor p21, 16 retinoblastoma (Rb) family member p130, 17 and FOXO1, 18 a transcription factor that upregulates expression of p27. 19 Recent studies have found that Skp2 also targets tumor suppressors involved in various cellular pathways: DNA double-strand break repair protein BRCA2 20 ; H3K4 methyltransferase MLL 21 ; Ras-association domain family protein (RASSF1A), a negative Ras effector 22 ; TIS21/BTG2/PC3, a downstream mediator of p53 signaling 23 ; and proapoptotic ING3. 24 While the net effect of Skp2-mediated ubiquitination supports cellular proliferation, SCFSkp2 was also reported to control degradation of oncoproteins, namely MEF/ELF4, a transcriptional activator that promotes S phase entry and proliferation, 25 and human papillomavirus (HPV) E7, which targets Rb for ubiquitin-mediated degradation (diagrammed in Figure 1). 26
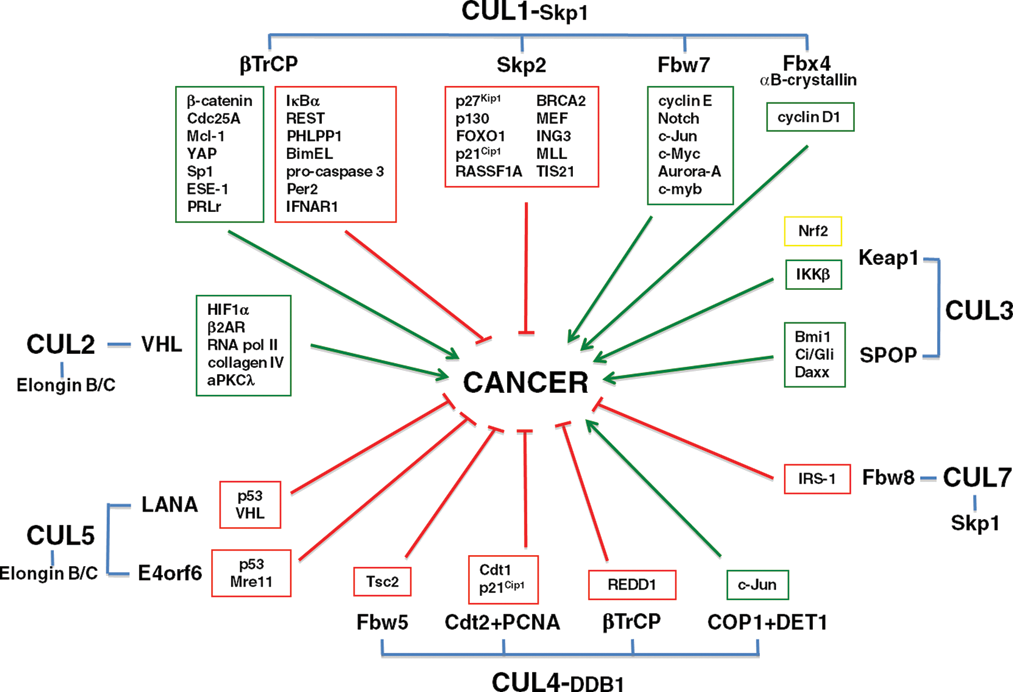
Schematic diagram of cullin/adaptor complexes, substrate receptors, and substrates. The roles of cullin substrates in promoting (green boxes/arrows) or inhibiting (red boxes/arrows) growth or survival, thus impacting oncogenesis, are indicated.
Proper regulation of Skp2 activity is vital for orderly entry into S phase, as elevated Skp2 expression may overcome p53-dependent cell cycle checkpoints. 27 For example, deficiency of the PTEN tumor suppressor leads to higher Skp2 levels and correspondingly reduced p27 stability. 28 In addition, the Akt signaling pathway may exert its proto-oncogenic effect in part through phosphorylation of Skp2, resulting in its relocalization to the cytoplasm, which impairs anaphase promoting complex (APC)–mediated Skp2 ubiquitination and destruction. 29,30 Thus, dysregulation of the PI3K/Akt signaling pathway may lead to the accumulation and enhanced activity of Skp2, and accelerate the turnover of its tumor suppressor substrates.
CUL1-βTrCP
While SCFSkp2 has a well-characterized role in the promotion of oncogenesis, the role of F-box protein βTrCP in maintaining the balance between normal and malignant growth is more complex and likely dependent on cell context. Mammals express two βTrCP isoforms, βTrCP1 and βTrCP2/HOS. The βTrCP1 knockout mouse is viable but exhibits defects in spermatogenesis and centrosomal overduplication 31 or shows no obvious phenotypic defects. 32 A number of SCFβTrCP substrates have been identified, and the meiotic and mitotic defects have been attributed to stabilization of Emi1, an SCFβTrCP substrate and inhibitor of the APC/C ubiquitin ligase. 31,33
SCFβTrCP potentially exerts prosurvival and oncogenic effects through the targeting of tumor suppressors that contain the conserved DSGXXS destruction motif. 34,35 Degradation of IκBα by SCFβTrCP relieves inhibition of NF-κB signaling and promotes cell cycle progression and survival, 36 which may also contribute to mammary tumorigenesis induced by enforced βTrCP1 expression in MMTV-βTrCP1 transgenic mice. 37 Interestingly, mutational activation of BRAF is frequently observed in human malignant melanomas and is coupled with enhanced βTrCP expression, constitutive induction of NF-κB activity, and increased survival of melanoma cells. 38 The prosurvival function of βTrCP is further substantiated by its selective targeting of the proapoptotic proteins procaspase 3 and BimEL for ubiquitin-mediated proteolysis. 39,40 Recent studies further identified new oncogenic roles for βTrCP in the degradation of REST, a tumor suppressor that promotes neuronal differentiation, 41 and PHLPP1, an inhibitor of Akt signaling. 42 Lastly, silencing of βTrCP suppresses proliferation and survival of breast cancer cells and augments the cytotoxicity of chemotherapy drugs. 43 Thus, targeting βTrCP may be beneficial for enhancing the efficacy of anticancer therapies.
However, SCFβTrCP also plays a significant role in restricting cellular levels of oncoproteins such as β-catenin. Dysregulation of β-catenin levels has been linked to both familial and spontaneous human colon cancer (reviewed in Phelps et al. 44 ). Mutations in βTrCP or the DSGXXS degron of β-catenin both resulted in increased accumulation of β-catenin, thus contributing to the pathogenesis of gastric and endometrial cancers. 45,46 Transgenic mice overexpressing either wild-type βTrCP or the dominant-negative ΔF-box–βTrCP in the intestine, liver, and kidney develop tumors in all targeted organs, in part through β-catenin activation. 47 Noubissi et al. showed that β-catenin signaling activates the expression of mRNA-binding protein CRD-BP (coding region determinant–binding protein), which in turn binds to the coding region of βTrCP, leading to its stabilization and elevated SCFβTrCP E3 ligase activity. 48 β-catenin overexpression also induces βTrCP-mediated degradation of Per2, a circadian clock protein with tumor suppressor activity. 49 Metabolic stress, such as glucose starvation, upregulates βTrCP expression to target the Sp1 transcription factor in prostate cancer cells, which suggests the possible therapeutic strategy of using small molecule agents that mimic glucose starvation. 50 Additional oncoproteins that are recognized by βTrCP following their phosphorylation include Mcl-1, 51 ESE-1, 52 and YAP. 53
In response to DNA damage, βTrCP plays a critical role in damage checkpoint activation by targeting Cdc25A and securin for degradation, thereby arresting the cell cycle and allowing time for DNA repair. 54 Furthermore, checkpoint termination and recovery from cell cycle arrest require proteolytic removal of Claspin, which is required for Chk1 activation by acting as an adaptor that bridges Chk1 and ATR. 55,56 Following DNA repair, Claspin is phosphorylated by the Plk1 kinase, which triggers its subsequent destruction by SCFβTrCP. 57-59 As such, βTrCP is a critical component of the DNA damage checkpoint pathway that ensures genomic integrity through coordination of the cell cycle and DNA damage checkpoint. Failure of βTrCP-mediated proteolysis may disrupt proper control of the DNA damage response. Moreover, given the demonstrated role of Cdc25A in promoting mammary tumorigenesis, 60 SCFβTrCP is anticipated to negate its tumorigenicity by targeting Cdc25A for proteasomal destruction.
In addition to cytosolic substrates, βTrCP also targets membrane proteins, such as the interferon receptor IFNAR1, which mediates type I IFN signal transduction. 61 IFN signaling is often downregulated in spontaneous tumors through multiple mechanisms, including aberrant IFNAR1 destruction by SCFβTrCP, and may account for the compromised efficacy of IFN treatment seen with human malignant melanomas. 62 Consistently, inhibition of βTrCP was shown to sensitize human melanoma cells to the antiproliferative effects of type I IFN, 63 suggesting a potential improvement of therapeutic efficacy by blocking βTrCP-mediated IFNAR1 proteolysis. Moreover, βTrCP also targets the prolactin receptor (PRLr) for ubiquitin-mediated endocytosis and lysosomal proteolysis. 64 PRLr degradation is impaired in breast tumor cells and tissues, which directly correlates with enhanced expression of PRLr in malignant breast epithelia. 65
Overall, βTrCP has been shown to both suppress and aid tumorigenesis, and this functional disparity of SCFβTrCP may be attributed to tissue-specific differences in signaling, cell cycle progression, and survival. Therefore, the role of βTrCP as an anticancer therapeutic target warrants further investigation.
CUL1-Fbw7
The Fbw7/Cdc4 substrate receptor plays a straightforward role as a tumor suppressor, as SCFFbxw7 ubiquitinates multiple substrates that promote growth and oncogenesis, including cyclin E (+Cdk2), 66 Notch, 67 c-Myc, 68,69 c-Jun, 70 Aurora-A, 71 c-myb, 72 p18-cyclin E, 73 and cyclin E2. 74 Knockout of Fbw7 in mice is embryonic lethal due to dysregulation of multiple substrates. 67,75 Fbw7 is a direct transcriptional target of p53 that is induced in response to genotoxic stress, thus triggering cell cycle arrest following SCFFbw7-mediated cyclin E degradation. 76
Fbw7 mutations or loss of heterozygosity have been found in numerous cancer types including breast, endometrial, pancreatic, and gastric cancers (reviewed in Welcker and Clurman 77 ). Moreover, mutations in the Fbw7 substrate-binding region have been found in over 30% of pediatric T-cell acute lymphoblastic leukemia patients and result in aberrant Notch signaling. 78 Dysregulation of Fbw7 activity may also result from mislocalization or inhibition through viral hijacking and interfere with the degradation of proto-oncogenic substrates. 79-81 Thus, Fbw7 is a bona fide tumor suppressor that plays vital roles in restraining proliferative signaling pathways and transcription programs.
CUL1-Fbx4
The F-box protein Fbx4 requires αB-crystallin as a coreceptor for targeted degradation of cyclin D1 in the cytoplasm. 82 Knockdown of Fbx4 or αB-crystallin resulted in the neoplastic transformation of mouse fibroblasts, indicating an anti-oncogenic role for this ubiquitin ligase. 83 Moreover, phosphorylation-dependent Fbx4 dimerization is required for cyclin D1 degradation, and mutations that inhibit dimerization have been found in human esophageal carcinomas. 83 SCFFbx4 also plays a role in telomere maintenance through the degradation of Pin2/TRF1. 84 Pin2/TRF1 induces apoptosis of cells with short telomeres in a cell cycle–dependent manner and is frequently downregulated in breast tumors. 85 Thus, the role of SCFFbx4 in blocking tumorigenesis is predominantly mediated by cyclin D targeting, while further studies are required to clarify its role in telomere maintenance in relation to oncogenic transformation.
CUL2 and CUL5
Both CUL2- and CUL5-based ubiquitin ligases assemble functional complexes using Elongin B/C as an adaptor to recruit substrate receptors that contain a suppressor of cytokine signaling (SOCS) box. To date, knockout models for Cul2 and Cul5 have not been published. In C. elegans, silencing of CUL2 by RNAi revealed that CUL2 is an essential gene for mitotic germline proliferation, meiotic division II following fertilization, and positioning of the anterior-posterior axis, 86-89 while CUL5 is not essential for growth or development. 90
The most extensively studied CUL2 substrate receptor is the von Hippel-Lindau (VHL) tumor suppressor, which targets the oxygen-sensing transcription factor HIF1α. 91 Mutations of the VHL tumor suppressor gene are causal for the familial von Hippel-Lindau disease, which is characterized by tumors of the eye, brain, spinal cord, kidney, pancreas, and adrenal glands. Analogous to Ser/Thr phosphorylation-dependent substrate recognition by F-box proteins of the CUL1 machinery, hydroxylation of specific proline residues on HIF1α by the PHD family of prolyl-4-hydroxylases creates a binding site for VHL and allows ubiquitination and proteasome-mediated degradation to occur under normoxic conditions. 92,93 Under hypoxic conditions, HIF1α accumulates and activates expression of proangiogenic genes such as VEGF and PDGF, leading to increased oxygen delivery by stimulating angiogenesis as well as oxygen utilization, increased transport of glucose and conversion to pyruvate, and reduction of mitochondrial mass. Further contributing to the tumor suppressive role of CUL2, VHL also targets activated atypical PKCλ, a regulator of cell polarity and growth, for ubiquitination. 94 β2-adrenergic receptor (β2AR) was recently identified as another substrate of VHL, and its hydroxylation by EGLN3, an O2-dependent iron-containing prolyl hydroxylase, is responsible for triggering β2AR ubiquitination and proteasomal degradation. 95 RNA polymerase II and collagen IV were also shown to interact with VHL in a prolyl hydroxylase–dependent manner, 96,97 suggesting that VHL likely targets a broad range of substrates in response to O2-dependent hydroxylation. Future studies will determine whether the defective degradation of additional CUL2-VHL substrates plays a role in the pathogenesis of von Hippel-Lindau disease.
HPV promotes oncogenic transformation by hijacking cellular ubiquitin ligases to target tumor suppressors. Notably, the HPV16 E6 oncoprotein diverts the specificity of the E6AP ubiquitin ligase to degrade p53. 98,99 In a similar manner, the HPV16 E7 oncoprotein redirects the CUL2 ubiquitin ligase to target the Rb tumor suppressor for destruction. 100 These studies highlight the paradigm of viral hijacking of the cellular ubiquitination machinery to destroy tumor suppressors, thereby generating a permissive environment for the DNA tumor virus to infect host cells and leading to cellular transformation.
While both CUL2- and CUL5-based ubiquitin ligases utilize Elongin B/C as adaptors for substrate recruitment, the two cullins appear to recruit distinct SOCS-box proteins as substrate recognition components. To date, no endogenous substrate receptors have been described for CUL5, but viral proteins were found to serve as substrate recognition components for CUL5-based ubiquitin ligases. Kaposi’s sarcoma–associated herpes virus (KSHV) encodes the latency-associated nuclear antigen (LANA), which is a SOCS-box substrate receptor for the CUL5–Elongin B/C complex that targets the tumor suppressors VHL and p53 for degradation. 101 In a similar manner, the HIV-1 viral infectivity factor (Vif) also contains a SOCS-box and assembles a CUL5-based ubiquitin ligase that mediates ubiquitination of the host antiviral factor APOBEC3G. 102 Alternatively, AAV E4orf6 utilizes BC-box motifs to interact with CUL5–Elongin B/C and direct degradation of p53 and Mre11. 103,104 Finally, CUL5 utilizes the Hsp90 chaperone complex instead of Elongin B/C to degrade ErbB2, 105 suggesting a physiological function of CUL5 in restricting cellular proliferation.
CUL3
CUL3-based ubiquitin ligases feature BTB domain–containing proteins that integrate the functions of both adaptors and substrate receptors (reviewed in Petroski and Deshaies 1 ). CUL3 is downregulated in breast and kidney cancer, suggesting a tumor suppressive role. 106 Conditional knockout of Cul3 in mice resulted in the accumulation of cyclin E and enrichment of cells in S phase, suggesting that CUL3 is essential to maintain quiescence in mammalian cells. 90,107
The best-characterized CUL3 substrate receptor is Keap1, which targets the Nrf2 transcription factor for ubiquitination and degradation. 108 In response to oxidative stress, Nrf2 upregulates transcription of endogenous antioxidant genes and phase II detoxifying enzymes. 109 Expression of Nrf2-dependent proteins is critical to moderate the effects of toxins and carcinogens, and maintain cellular redox homeostasis. Although Nrf2 itself is not essential for development, Nrf2 knockout mice exhibit an impaired response to oxidative stress, resulting in inflammatory disorders of multiple organs 110 (reviewed in Kensler et al. 111 ). Since chronic inflammation may contribute to carcinogenesis by appropriating signaling pathways that support growth (reviewed in Lu et al. 112 ), the Nrf2-triggered antioxidant response must be precisely controlled and can be further exploited as a potential target for cancer prevention. Keap1 knockout in mice is lethal postnatally and is likely due to Nrf2 accumulation and constitutive activity. 113 Nrf2 knockout rescued the postnatal lethal phenotype of Keap1 knockout mice, suggesting that Nrf2 is the major target of the CUL3-Keap1 ubiquitin ligase. 113 Consequently, CUL3-mediated degradation of Nrf2 is highly significant in the cellular antioxidant and stress response that promotes the survival of both normal and cancer cells.
Tumors from approximately 19% of lung cancer patients harbor somatic mutations in Keap1 that prevent effective Nrf2 ubiquitination, indicating an anti-oncogenic role for Keap1. 114 Conversely, two Nrf2 mutation hot spots were recently identified in approximately 10% of lung cancer patients that enable the transcription factor to evade Keap1-mediated degradation. 115 There is also increasing evidence that the Nrf2-dependent antioxidant effect can be co-opted by cancer cells. Nrf2 and its downstream genes are overexpressed in many cancer cell lines and human cancer tissues, conferring a survival and growth advantage (reviewed in Hayes and McMahon 116 ). Furthermore, Nrf2 is upregulated in chemoresistant cancer cells and is thought to be responsible for acquired chemoresistance. 114,117 Therefore, targeted inhibition of the Nrf2 pathway may enhance the efficacy of chemotherapy.
Another substrate of the CUL3-Keap1 ubiquitin ligase is IKKβ, which is involved in tumor development and progression through activation of the NF-κB pathway. 118 Keap1 knockdown resulted in the stabilization and accumulation of IKKβ and the subsequent upregulation of NF-κB–dependent angiogenic factors. Thus, dysregulation of CUL3-mediated IKKβ ubiquitination is likely a contributing factor to tumorigenesis.
The BTB protein SPOP also has been found to act as a substrate receptor for CUL3 and may function as a tumor suppressor through the degradation of Daxx, a transcriptional repressor of p53. 119 Moreover, CUL3-SPOP was found to target Bmi1, 120 an oncoprotein that is required for the self-renewal of hematopoietic stem cells. 121 CUL3-SPOP also regulates Hedgehog signaling through ubiquitination of the Ci/Gli family of transcription factors. 122 Aberrant Hedgehog signaling and Ci/Gli activity have been frequently found in basal cell carcinomas (reviewed in Yang et al. 123 ), and SPOP-mediated control of Ci/Gli protein stability likely represents an important regulatory mechanism to control this critical signaling pathway.
CUL4A and CUL4B
The CUL4 family has two members, CUL4A and CUL4B, that share extensive sequence homology and functional redundancy in maintaining growth and survival. While other cullins utilize BTB-fold–containing adaptors (Skp1 or Elongin B/C) to bind their substrate receptors, the CUL4 family employs the structurally distinct triple WD40 domain–containing DDB1 adaptor to recruit the DCAF (DDB1-CUL4–associated factors) family of substrate receptors 124-127 (reviewed in Lee and Zhou 128 ).
The CUL4A gene was initially found to be amplified or overexpressed in primary breast cancer. 129 Recent genome-wide analysis of human cancers by cGH identified CUL4A amplification in 5% of familial and sporadic breast cancers, and as high as 20% of the basal-like breast cancer subtype that is characterized as “triple negative” (ER, PR, and HER2/Neu) and associated with aggressive growth and poor prognosis. 130 CUL4A amplification has also been reported in squamous cell carcinomas, 131 adrenocortical carcinomas, 132 childhood medulloblastoma, 133 hepatocellular carcinomas, 134 and approximately 64% of primary malignant pleural mesothelioma tumors. 18 Moreover, patients with high CUL4A expression have significantly shorter overall and disease-free survival. 135 Dysregulation of CUL4A in multiple tumor types leads to the hypothesis that CUL4A plays a role in promoting oncogenesis. Mouse models support this scenario, as conditional Cul4a knockout in the skin caused a marked resistance to UV-induced carcinogenesis compared to wild-type and heterozygous mice. 136 To date, a Cul4b knockout mouse has not been reported, and the role of CUL4B in carcinogenesis remains to be determined.
The heterodimeric DDB1-DDB2 DNA damage sensors initiate the nucleotide excision repair (NER) pathway following UV irradiation, and were among the first identified targets of CUL4A ubiquitin ligase activity. 137-139 Follow-up studies revealed that DDBs are also integral components of the CUL4 ubiquitin ligase. 140 Mutations of DDB2 have been found in xeroderma pigmentosum group E patients and are causal for their photosensitivity and greatly increased skin cancer rates. 141 DDB2 acts as a substrate receptor in complex with CUL4A and DDB1 to target yet another NER damage sensor, XPC, for ubiquitination that increases its affinity for DNA. 142 Downregulation of DDB2 and its substrates limits NER activity, while enforced expression of DDB2 or knockout of Cul4a results in greater DNA repair following genotoxic insults. 136,143
CUL4A also plays a significant role in cell cycle regulation as Cdt2, another CUL4A substrate receptor, that targets Cdt1 and p21 for ubiquitin-proteolytic degradation with PCNA acting as an additional cofactor. 127,144-146 Both proteins regulate entry into S phase, with Cdt1 acting as a replication licensing factor and p21 acting as a cyclin-Cdk inhibitor. Knockdown of Cdt2 results in G2 arrest and DNA rereplication of the genome, 111 indicating a critical role for CUL4-Cdt2 in limiting the replication of DNA during S phase. In response to UV or ionizing radiation, Cdt1 and p21 are both rapidly degraded in a CUL4A-dependent manner. 144,147,148 Protein levels of p21 are higher in Cul4a knockout mice following UV irradiation and may contribute to the antitumorigenic effect by prolonging G1 arrest to provide time for the completion of NER activities. 149 CUL4B also plays a role in cell cycle progression, as knockdown in tumor cells resulted in cyclin E accumulation and cell cycle arrest in S phase. 150,151
The role of CUL4A as an oncogene is further supported by the recent finding that Merlin, a tumor suppressor encoded by NF2, inhibits the ubiquitin ligase activity of CUL4A in complex with VprBP/DCAF1. 152 Furthermore, mutations in Merlin that disrupt binding to CUL4A-VprBP and lead to a loss of enzymatic inhibition have been observed in neurofibromatosis patients. 152 Silencing of VprBP compromised tumorigenic activity in Merlin-deficient mesothelioma cell lines, indicating that Merlin may exert its anti-oncogenic effect through the inhibition of CUL4A-VprBP ubiquitin ligase activity. 152 VprBP is required for cell cycle progression into S phase, and HIV Vpr hijacking of the CUL4A-VprBP complex results in G2 arrest. 153-157 However, the cellular targets of VprBP have yet to be identified. Taken together, these findings indicate a role for the CUL4-VprBP ubiquitin ligase in promoting cell cycle progression and oncogenic transformation.
A CUL4A-based ubiquitin ligase featuring the Fbw5 substrate receptor regulates tuberous sclerosis protein 2 (Tsc2) turnover. 158 Tuberous sclerosis is an autosomal-dominant disease that is marked by the formation of hamartomas, or benign growths, on the skin, nervous system, kidneys, and heart. Mutations in the Tsc1 and Tsc2 tumor suppressors are causal for the disease through the loss of their negative regulation of the mTOR pathway. Katiyar et al. recently reported that CUL4A also targets REDD1, another inhibitor of mTOR signaling, with βTrCP utilized as the CUL4A substrate receptor. 149 Thus, CUL4A may exert its oncogenic effect in part by promoting mTOR signaling.
Contrary to the trend of CUL4A-mediated degradation of tumor suppressors, the c-Jun proto-oncogene is targeted for proteolysis by the COP1 substrate receptor. 6 CUL4A also regulates granulopoiesis by degrading the HOXA9 homeodomain protein, with the potential to restrict HOXA9-induced leukemogenesis. 159 Moreover, the NUP98-HOXA9 fusion protein, derived from the t(9;11)(p15;p15) chromosomal translocation in acute myeloid leukemia patients, is resistant to CUL4A-mediated degradation, which contributes to its potent leukemogenic activity. 160 In addition to restricting the NER threshold, CRL4 also participates in the cellular response to DNA damage through the ubiquitination of histones H3 and H4, which facilitates the recruitment of repair proteins (e.g., XPC) to damaged DNA. 161 Therefore, the CUL4A E3 ligase may play distinct roles in tumorigenesis that are dictated by the cellular context or environmental conditions.
CUL7
Least is known about how CUL7 assembles functional ubiquitin ligase complexes, as a clear paradigm has not been defined. To date, two F-box proteins have been identified as substrate receptors for CUL7, indicating a similar mode of assembly as CUL1-based ubiquitin ligases. Mutations in CUL7 are causal for 3M syndrome, an autosomal-recessive disorder marked by severe prenatal and postnatal growth retardation. 162 Cul7 knockout mice are runted with vascular defects, 163 indicating a vital role for CUL7 in cell growth and vascular morphogenesis. Additionally, CUL7 mRNA is significantly overexpressed in non–small cell lung carcinoma and is associated with poor patient prognosis. 164
CUL7 is a putative tumor-promoting protein, as it binds directly to the p53 tetramerization domain and antagonizes p53 function. 165 Knockdown of CUL7 results in the elevation of p53 protein levels, and CUL7 cooperates with myc transformation by inhibiting myc-induced p53-dependent apoptosis. 164 CUL7 assembles an SCF-like complex containing Skp1, Rbx1, and Fbw8 to target insulin receptor substrate 1 (IRS-1) for degradation, which may result in the negative regulation of mTOR signaling. 166 Similar to Cul7 knockout animals, Fbw8-null embryos are smaller than their wild-type and heterozygous littermates, with elevated insulin-like growth factor–binding protein 1 (IGFBP1). 167 Thus, the CUL7-Fbw8 complex plays a significant role in growth control through the regulation of insulin signaling.
Perspective
Although cullins have been shown to target multiple substrates with diverse functions, there are dominant substrates that, in a specific cellular context, dictate the contribution of the cullin in tumorigenesis. Nevertheless, a significant bottleneck in the ubiquitin field is the identification of substrates, as many putative substrate receptors have been identified, but their corresponding substrates remain to be determined. The assignment of receptors with their substrates will shed light on cullin-mediated mechanisms of oncogenesis. There is also increasing evidence that the regulatory mechanisms underlying the expression or posttranslational modifications of cullins may be exploited by cancer cells or DNA tumor viruses.
The intimate involvement of cullins in tumorigenesis indicates the cullin-mediated ubiquitination pathways as attractive targets for intervention and prevention of carcinogenesis. In this regard, proteasome inhibitors are currently used to treat multiple myeloma (reviewed in Shah and Orlowski 168 ). Moreover, an anti-neddylation agent was shown to restrict cullin E3 ligase activity. 169 However, more specific inhibitors, such as those that limit the activity of specific cullins or receptor-substrate complexes, are necessary to reduce the pleiotropic effects that are associated with nonselective “pan-cullin” or proteosome inhibition.
Given the multisubunit nature of cullin ubiquitin ligases and the extensive interfaces for protein-protein interactions, devising competitive inhibitors that target a specific binding interface between cullin subunits may appear technically challenging at first glance. However, the strict requirement for the subunits not only to assemble together but also to orient the substrate in close proximity to the E2 enzyme to facilitate ubiquitin transfer may provide additional intervention points for allosteric inhibitors. RNAi-based agents offer an alternative strategy to block the E3 ligase activity of specific cullins or substrate receptors at the biosynthesis level. Additionally, manipulation of cullin activity may be utilized for cancer prevention or therapy. In a manner similar to tumor virus hijacking of cullins to target the degradation of tumor suppressors, cullin specificity may also be manipulated through engineering known binding domains of substrate receptors to target oncogenes for degradation. 170-173 With increasing interest from both academia and the pharmaceutical industry in exploiting the ubiquitin pathwayas a target for intervention, there is a high likelihood for the ubiquitome to become an attractive source for the discovery and development of new therapeutics against cancer and other human diseases.
Footnotes
Acknowledgements
The authors thank Jeffrey Hannah for critical reading and editing of the paper. They apologize to all colleagues whose papers were not cited in this review due to space constraints.
The authors’ work is supported by National Institutes of Health (NIH) grants R01 CA098210 and CA118085. P.Z. is supported by the Irma T. Hirschl Career Scientist Award and the STARR Cancer Consortium.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.