
Abstract
Hematopoiesis is a process capable of generating millions of cells every second, as distributed in many cell types. The process is regulated by a number of transcription factors that regulate the differentiation along the distinct lineages and dictate the genetic program that defines each mature phenotype. Myc was first discovered as the oncogene of avian leukemogenic retroviruses; it was later found translocated in human lymphoma. From then on, evidence accumulated showing that c-Myc is one of the transcription factors playing a major role in hematopoiesis. The study of genetically modified mice with overexpression or deletion of Myc has shown that c-Myc is required for the correct balance between self-renewal and differentiation of hematopoietic stem cells (HSCs). Enforced Myc expression in mice leads to reduced HSC pools owing to loss of self-renewal activity at the expense of increased proliferation of progenitor cells and differentiation. c-Myc deficiency consistently results in the accumulation of HSCs. Other models with conditional Myc deletion have demonstrated that different lineages of hematopoietic cells differ in their requirement for c-Myc to regulate their proliferation and differentiation. When transgenic mice overexpress c-Myc or N-Myc in mature cells from the lymphoid or myeloid lineages, the result is lymphoma or leukemia. In agreement, enforced expression of c-Myc blocks the differentiation in several leukemia-derived cell lines capable of differentiating in culture. Not surprising, MYC deregulation is recurrently found in many types of human lymphoma and leukemia. Whereas MYC is deregulated by translocation in Burkitt lymphoma and, less frequently, other types of lymphoma, MYC is frequently overexpressed in acute lymphoblastic and myeloid leukemia, through mechanisms unrelated to chromosomal translocation, and is often associated with disease progression.
Overview of Hematopoiesis
Hematopoiesis is a process capable of generating 300 millions of cells per minute in the bone marrow of an adult human. These cells belong to many cell types, which widely differ in biological functions and number in the blood. All these morphologically, phenotypically, and functionally distinct cell types arise from pluripotent hematopoietic stem cells (HSCs) that are capable of self-renewal and differentiation through life. 1,2 The self-renewal and quiescence of hematopoietic stem cells are controlled by a highly orchestrated integration of environmental signals, most of which originate from the stem cell niche. 3–5 The so-called long-term HSCs (LT-HSCs) have the ability to self-renew and sustain hematopoiesis. 4 This fraction of HSCs remains largely quiescent or even dormant throughout the lifetime of an organism. LT-HSCs differentiate into multipotent progenitors (MPPs), which are cells with diminished self-renewal that can give rise to common lymphoid progenitors (CLPs) and common myeloid progenitors, which in turn differentiate to produce T- or B-lymphocyte precursors and granulocyte/monocyte precursors (GMPs) or megakaryocyte/ erythroid precursors (MEPs), respectively. 6 These multipotent progenitors undergo several steps of differentiation that give rise to committed progenitors and, ultimately, the diverse mature blood cells (Figure 1). The myeloid lineage includes erythrocytes, megakaryocytes producing platelets, different subclasses of granulocytes (neutrophils, eosinophils, basophils), monocytes–macrophages, and mast cells. The lymphoid lineage consists of T cells, B cells, and natural killer cells. Dendritic cells can be derived from either the myeloid or lymphoid pathway.
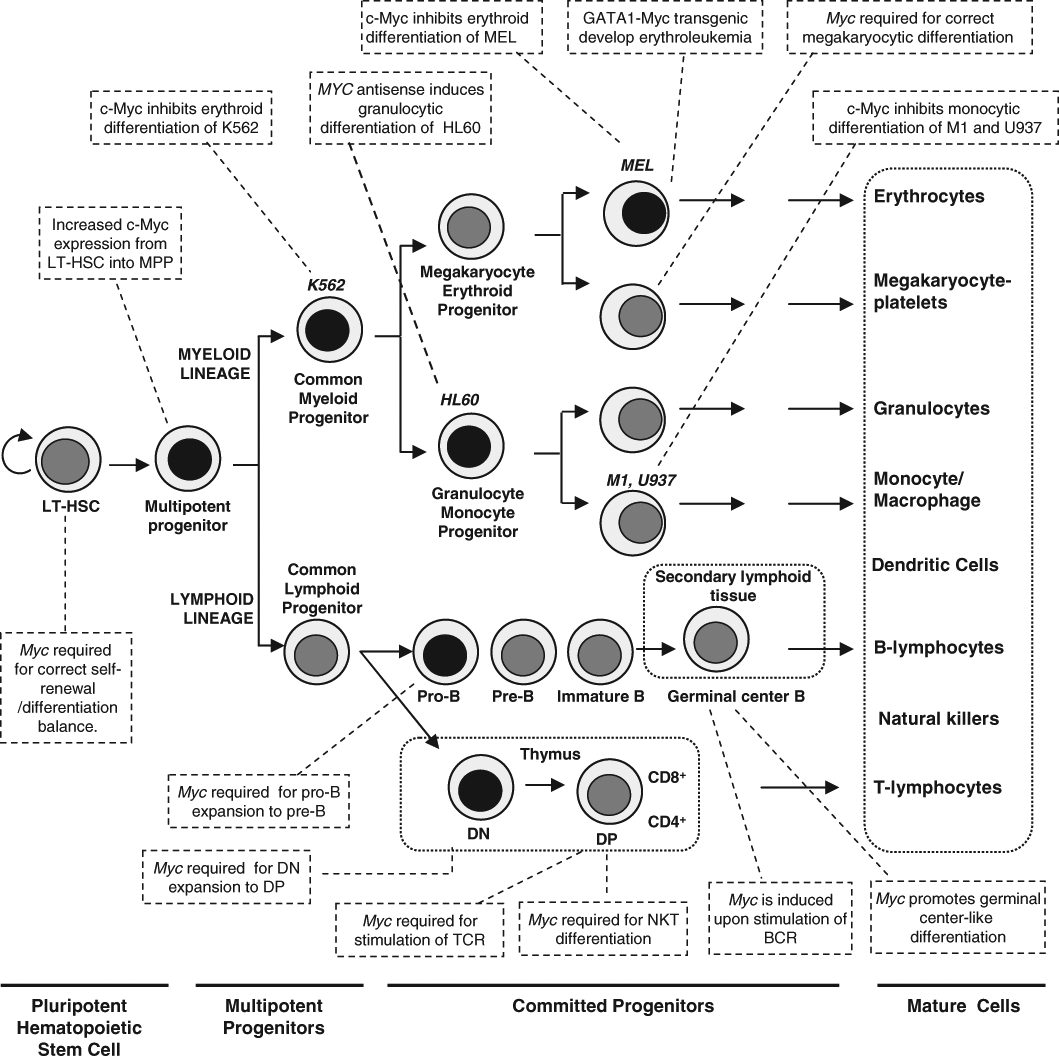
Classical model of hematopoiesis and the involvement of c-Myc in the process. Hematopoiesis begins at the level of pluripotent hematopoietic stem cells, which diverge into myeloid and lymphoid multipotent progenitors. These cells differentiate into committed progenitors and, finally, mature blood cells. T cells and B cells undergo further maturation in secondary lymphoid organs, such as spleen, lymph nodes, and mucosa-associated lymphoid tissues. Several leukemia-derived cell lines (MEL, K562, HL60, U937, M1), used as models to study c-Myc functions, are shown at their approximate stage of differentiation. Some reported effects of c-Myc on the differentiation of hematopoietic lineages are indicated in the boxes. Black nuclei indicate higher Myc expression. See the text for details and references. LT-HSC = long-term hematopoietic stem cells; MPP = multipotent progenitor; DN = double-negative T cell (CD4-, CD8-); DP = double-positive T cell; BCR = B-cell receptor; NKT = natural killer T cells.
Hematopoietic cell differentiation is controlled by intercellular and intracellular signaling mechanisms that target transcriptional regulators that in turn establish complex transcriptional networks. Therefore, lineage commitment relies on the timely activation of appropriate transcription factors and the silencing of inappropriate ones. 7 Comprehensive reviews have dealt with the transcriptional control in the development of erythroid, 8,9 myeloid, 10 B-cell, 11 and T-cell 12 lineages. Moreover, the hematopoietic system must respond to important loss of tissue (i.e., by bleeding after injuries) to replenish the lost cells in the appropriate relative numbers. This means that hematopoiesis must be exquisitely regulated by a large group of cytokines by cell–cell interactions in the bone marrow niche, and by the concerted activity of transcription factors. The data accumulated over recent years in cell and animal models indicate that c-Myc is one of the pivotal transcription factors regulating hematopoiesis. Figure 1 shows a schematic overview of hematopoiesis and the involvement of c-Myc in the process.
MYC Roles in Hematopoietic Cell Differentiation in Culture
Gene expression deregulation owing to chromosome aberrations or epigenetic alterations in hematopoietic stem cells or multipotent progenitors gives rise to different hematopoietic malignancies. Immortalized cell lines derived from leukemias or lymphomas have been extensively used as suitable model systems to study hematopoietic cell differentiation. Myeloid leukemia cell lines are arrested at different stages of maturation and can be induced to differentiate into several pathways that resemble normal counterparts, depending on their differentiation potential (e.g., multipotent K562 cells, bipotent HL60 cells, or unipotent U937 cells; see Figure 1). These hematopoietic models, among others, have helped to highlight the importance of c-Myc on cellular processes such as proliferation, differentiation, and apoptosis.
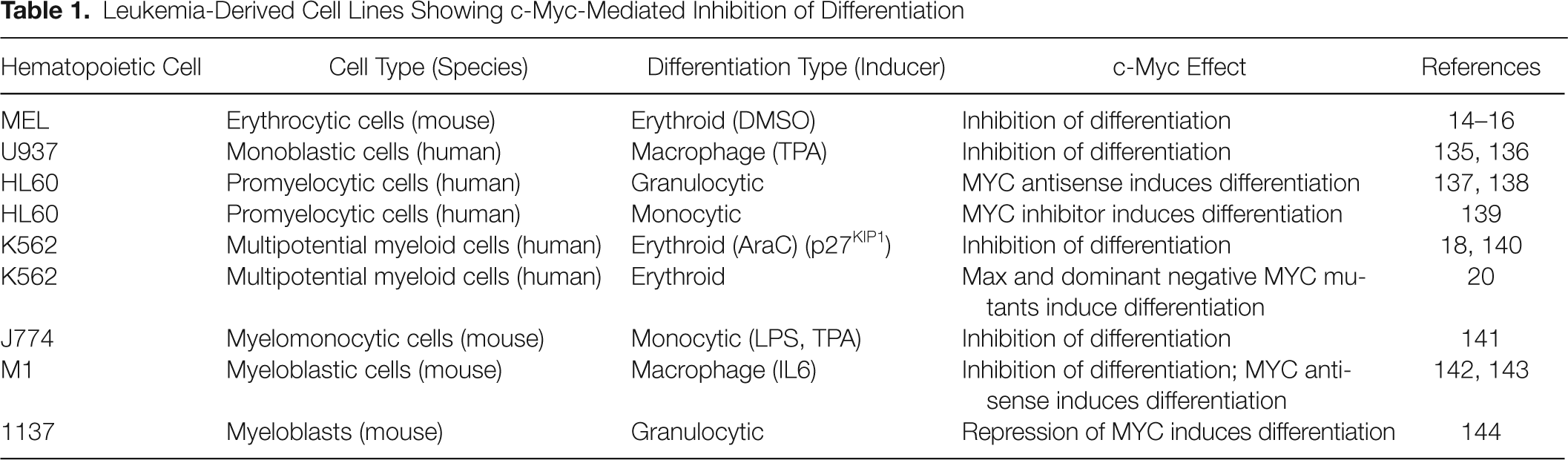
Inhibition of cell differentiation was one of the first biological effects described for c-Myc (reviewed in Reference 13). In 1986, three reports showed that c-Myc blocked the chemically induced erythroid differentiation of Friend murine erythroleukemia cells. 14–16 In the same year, it was reported that B-lymphocyte development was impaired in young Eµ-Myc transgenic mice before the onset of lymphoma. 17 Since then, c-Myc ectopic expression has been found to inhibit differentiation in a number of cell lines and primary cells, and about half of them are hematopoietic cell lines (Table 1). However, inhibition of differentiation is not a universal c-Myc activity. For instance, in the tripotent cell line K562, we found that whereas c-Myc impairs AraC-induced erythroid differentiation, 18 it does not impair TPA-induced myelomonocytic differentiation or staurosporine-induced megakaryocytic differentiation. 19 Consistent with those results, c-Myc-dominant inhibitory mutants enhance erythroid but not megakaryocytic differentiation. 20
Leukemia-Derived Cell Lines Showing c-Myc-Mediated Inhibition of Differentiation
In genetically defined models where erythroid differentiation is induced by p27KIP1, c-Myc blocks differentiation. 21 In this model, c-Myc impairs the upregulation of many erythroid-specific genes, as well as that of transcription factors that determine erythroid lineage differentiation (including GATA1 and NFE2); but, strikingly, it does not reverse the proliferation arrest and the repression of CDK activity mediated by p27KIP1. This suggests that c-Myc operates through the regulation of several or many genes, which is to be expected in view of the broad changes in the gene expression profile induced by c-Myc. In a complementary approach using the leukemia K562 and U937 cells, it was shown that c-Myc is not downregulated when cells are growth arrested but not differentiated. 22,23 All together, the available data indicate that c-Myc can block differentiation of hematopoietic cell models in culture through mechanisms distinct from those involved in cell cycle progression.
MYC Expression in Hematopoietic Progenitors and HSCs
The involvement of c-Myc in hematopoiesis regulation was soon suggested by 3 avenues: First, all three oncogenic Myc retroviruses originally isolated induced chicken hematopoietic tumors, which were a type of myeloid leukemia (myelocytomatosis) 24 ; second, MYC deregulation was first observed in Burkitt lymphoma as well as in chemically induced murine plasmacytomas 25 ; and, third, inhibition of hematopoietic cell differentiation in culture was one of the first biological effects of c-Myc. Later on, the development of several mice models with targeted mutation of c-Myc confirmed its important role in hematopoiesis. We briefly review the conclusions drawn from several of these models.
There is a differential expression of Myc family members in hematopoietic lineages: c-Myc and N-Myc transcripts are coexpressed at similar levels in LT-HSCs 26 and are detected in most progenitor subsets. In contrast, L-Myc mRNA is not expressed in any stem/progenitor cells, and it is only modestly expressed in CLPs, megakaryocytes, and macrophages (3%–5% of total Myc). Note that no hematopoietic cell type expresses only N-Myc or L-Myc without c-Myc. 26 The pervasive c-Myc expression in the hematopoietic system seems to be in correspondence with the absolute prevalence of c-Myc deregulation in human leukemia and lymphoma, as compared with the other family members (see below).
The expression of c-Myc at the protein level in hematopoietic progenitors and mature lineages was studied in a mouse line where the endogenous Myc locus was replaced with an allele encoding a GFP-c-Myc fusion protein. These Myc G/G mice were viable and showed an apparently normal hematopoiesis. 27 The highest expression of c-Myc (i.e., c-Myc–GFP in this model) was detected in the myeloerythroid progenitor fraction of the adult bone marrow, a cell population that originates directly from Lin−Sca1+c-Kit+ (LSK) cells and is actively proliferating. 28 Adult LSK cells include LT-HSCs and MPPs. 4,5 Interestingly, those LSK cells with lower c-Myc expression (i.e., GFP expression) corresponded to LT-HSCs (the more primitive and undifferentiated population), whereas the LSK expressing more c-Myc corresponded to MPPs. 28 Note that the abundance of c-Myc protein was substantially greater in fetal liver LSK cells than in adult LSK cells, 28 which seems in concordance with the fact that fetal liver HSCs actively proliferate during embryonic days 12.5 to 14.5. 29 Interestingly, Myc mRNA is already expressed at significant levels in LT-HSCs, but the increase in c-Myc protein expression (i.e., c-Myc-GFP) from LT-HSCs into MPPs is not observed at the mRNA level. 26 This suggests that the protein changes are due to posttranscriptional mechanisms, which complicates the interpretation of previous data obtained measuring mRNA levels.
MYC Roles in Hematopoietic Stem Cells: Lessons from Targeted Mutation in HSCs in Mice
Mice embryos in which both Myc alleles have been mutated by gene targeting in ES cells fail to thrive and die before midgestation (embryonic day ~10.5). Myc-deficient embryos are much smaller and exhibit several defects, with the impaired hematopoiesis and angiogenesis being the most prominent and reproduced in different models. 30–32 Whether deletion of Myc impairs vasculogenesis is controversial. 30,33 There are data that strongly suggest that defective hematopoiesis is the main reason for Myc-dependent embryonic lethality of Myc-deficient embryos. First, Myc-deficient embryos survive up to embryonic day 10.5, coincident with the onset of fetal liver hematopoiesis. Second, when Myc deletion was generated in the epiblast (i.e., with wild-type placenta), it was shown that Myc is specifically required for yolk sac primitive and intraembryonic definitive hematopoietic development, although nonhematopoietic defects—such as abnormalities of the heart, pericardium, and neural tube; delay in embryo turning; and small embryo size—were virtually absent in these embryos. 33,34 Finally, embryos lacking Myc in hematopoietic lineages (deleted in cells expressing the hematopoietic gene Vav1) phenocopied those lacking Myc in the entire embryo, although they survived until embryonic day 11.5 to 12.5 (i.e., 1 to 2 days longer than the complete null embryo). 33
Although the above complete Myc null mouse demonstrates a role of c-Myc in hematopoiesis at an early stage, there is more controversy to the precise role of c-Myc in the process. This question has been addressed by conditional knockouts targeting Myc in hematopoietic precursors. The conditional deletion of Myc in the bone marrow (and most other tissues) achieved by the postnatal induction of the Cre gene in MxCre; Myc flox/flox mice results in severe cytopenia and accumulation of LT-HSCs. 35 It has been suggested that this accumulation is caused not by alterations in HSC self-renewal or survival but rather by a failure to initiate normal stem cell differentiation, likely caused by increased HSC–niche adhesion. 35
Consistent with the phenotype of Myc knockouts, enforced Myc expression in HSCs results in lower HSC numbers, 35 accompanied with reduced expression of N-cadherin and integrins. In this scenario, high c-Myc levels would lead to detachment of the HSCs from their bone marrow niche, reduced self-renewal activity, and increased proliferation and differentiation of the Myc-expressing HSCs, thus exhausting the pool of LT-HSCs. 35 Murine HSCs (Lin−Sca1+ cells) retrovirally transduced with a Myc-ER gene consistently showed extended proliferation in culture when treated with the c-Myc activator (hydroxytamoxifen). 36 In agreement with this c-Myc effect, the bone marrow of mice deficient for p27KIP1 and Mxd1 (formerly Mad1) genes has an expanded pool of quiescent HSCs, which is consistent with the phenotype observed in Myc −/− mice owing to the well-known activity of both proteins as c-Myc antagonists. 37
Other investigators, using the same mice model, observed that c-Myc loss in adult bone marrow results in the transient accumulation of HSCs, followed by the progressive loss of bone marrow cells, which is maximal at 8 weeks after Myc deletion. 38 Perinatal deletion of Myc (again using the Mx-Cre system) results in the accumulation of a population of HSCs with the phenotype Lin-negative, Sca1+-c-Kit− cells (versus the c-Kit-positive found in wild-type HSCs). These cells are deficient in differentiation, and it is suggested that they reflect a potential primitive progenitor. 38
Myc-deficient HSCs produced by either perinatal or adult excision by the Cre-Myc flox/flox model fail to engraft in recipient mice in a competitive or noncompetitive setting. This demonstrates that the accumulated Myc-deficient HSCs are functionally defective, and the impaired hematopoiesis can be attributed to an intrinsic defect of the Myc-deficient cell rather than to an aberrant microenvironment in the bone marrow. 35,38
The results commented above refer to c-Myc effects in HSC biology. As already mentioned, both c-Myc and N-Myc mRNAs are expressed at similar levels in HSCs, so it is interesting to compare the effects of both Myc genes and their concomitant deletion in HSCs. The deletion of N-Myc alone does not seem to affect HSCs or hematopoiesis, but the simultaneous deletion of Myc and N-Myc results in HSCs that proliferate less than c-Myc−/− HSCs and undergo cell death, meaning that N-Myc plays a role in HSC survival. 26 Interestingly, replacement of Myc by N-Myc in mice results in normal development, hematopoietic, and lymphoid differentiation. 39
The c-Myc role in HSCs has been confirmed in mice with targeted mutation of the ubiquitin ligase Fbw7, which is the major (and, probably, the only) ubiquitin ligase for Myc in HSCs. 28 Furthermore, Fbw7 deletion in bone marrow and fetal liver HSCs results in elevated expression of c-Myc, accompanied with a reduced fraction of LT-HSCs. 28,40 This is fully consistent with the accumulation in LT-HSCs observed in Myc-deficient mice described above. 26,35,38 Moreover, concomitant Myc deletion rescues the effect of Fbw7 deficiency, expanding the number of HSCs. 28 Collectively, these investigations indicate that enforced c-Myc leads to loss of self-renewal activity at the expense of differentiation, whereas c-Myc deficiency results in increased HSC self-renewal and accumulation of HSCs. In summary, in the HSC population, c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation.
MYC in the Differentiation of Lymphoid Cells In Vivo
MYC involvement in the lymphoid lineage development has received much attention because of its heavy involvement in human lymphoma. Both N-Myc and c-Myc mRNA are expressed during the maturation and expansion of the earliest B-cell precursors (pro–B cells) into pre–B cells. Thereafter, only c-Myc is expressed in mature B cells after B-cell activation. 41–43 Myc expression consistently increases in response to B-cell receptor (BCR) stimulation in vitro. 44,45 In mice, pro–B cells and pre–B cells proliferate in response to cytokines such as IL-7 produced by bone marrow stromal cells, and this response is enhanced in pre–B cells from Eµ-Myc mice. 46
This is confirmed by measuring GFP-Myc protein in the lymphoid compartment of the GFP-Myc cell line described above. Flow cytometry studies showed that c-Myc expression is dynamically regulated in developing and mature B and T lymphocytes in vivo. Despite the broad cellular effects of c-Myc, one study showed a close correlation between GFP-c-Myc levels and proliferation, both in lymphocyte differentiation and upon activation of mature lymphocytes, suggesting that c-Myc is required to promote proliferation of lymphocytes. 27
As commented below, transgenic mice overexpressing MYC (the human ortholog) in B cells via the Eµ enhancer develop lymphoma with relatively long latencies. During the prelymphomatous state, the constitutive c-Myc expression results in a higher number of pre–B cells and a lower number of mature B cells, but the B cells are able to mature relatively normally despite the presence of deregulated c-Myc. 17
In contrast, mice deficient in c-Myc in early B-lymphocyte precursors (pro–B stage) have impaired B-cell differentiation (Moreno de Alboran, personal communication). Conditional deletion of Myc in mature B cells results in the impaired proliferation of Myc null B lymphocytes in response to CD40 and IL-4. 47 N-Myc expression from the Myc locus is capable of driving B- and T- lymphocyte differentiation in knock-in mice. 39 One step further, the conditional deletion of both c-Myc and N-Myc, using CD19-cre mice, inhibits B-cell development at the transition from pro–B cell to pre–B cell (i.e., the stage at which the combined signaling from the pre-BCR and IL-7 normally induce c-Myc and N-Myc expression). 46 The data indicate that Myc stimulates B-cell differentiation and expansion at least from the pre-BCR stage.
As in the case of B cells, c-Myc and N-Myc are expressed in immature T lymphocytes, but c-Myc is the only Myc gene expressed after the pro–T cell stage and in mature T cells. 27,41,43 In parallel with the B-cell scenario, c-Myc is upregulated upon T-cell receptor stimulation. 42,48
Based on 2 different models, it has been shown that Myc −/− T cells (in LckCre Mycflox/flox mice) cannot populate the adult thymus and that subsequent thymocyte maturation is ineffective: The cells fail to proliferate normally at the late double-negative CD4−CD8− stage, but this is not required for the progression from double-negative into double-positive (CD4+CD8+) cells. 42,49 However, conditional ablation of Myc in double-positive thymocytes (in CD4Cre Mycflox/flox mice) impairs natural killer T-cell (NKT) development arresting intrathymic NKT proliferation upon agonist selection, whereas conventional T-cell development is not affected. 50,51
In response to infections, human B lymphocytes transiently pass through the germinal center, where antigen-specific B cells suffer somatic hypermutation, class switch recombination, and clonal expansion. 52 In Burkitt lymphoma, which is the hallmark of Myc-dependent neoplasia in humans, B-cell differentiation into plasma cells is impaired. c-Myc overexpression in Burkitt lymphoma cells induces a constitutive centroblast-like phenotype in the ganglia germinal center, thus blocking the differentiation into mature B cells. This effect may be in part mediated by the MYC-dependent upregulation of the germinal center–specific transcription factor Bcl6. 53 Thus, c-Myc seems to impair the differentiation program as the primary cause of this lymphoma.
MYC in the Differentiation of Myeloid Cells In Vivo
The expression of Myc genes in the myeloid progenitors has merited less attention than that of lymphoid progenitors, but the effects of Myc deletion in the myeloid compartment have been recently reported in the c-Myc conditional knockout mice. 54 Besides the reduced lymphocyte number discussed above, adult c-Myc−/− mice show significant thrombocytosis, severe anemia, and grossly decreased neutrophil/monocyte number. 54 Thus, c-Myc induces opposite effects in the differentiation of megakaryocytic versus monocytic and erythroid lineages. Moreover, these effects of c-Myc deletion in vivo on hematopoietic lineages correlates well with c-Myc expression in mouse hematopoietic cells: Cells expressing higher levels of c-Myc, such as GMPs, CLPs, and erythrocytic blasts, are significantly reduced, whereas cells expressing lower Myc levels (HSCs, MEPs, and megakaryocytes) are less affected or are increased in number. 54 The data are roughly consistent with the observations in cell culture models where c-Myc impairs monocytic, granulocytic, and erythroid differentiation (see above and Table 1).
Megakaryocytes from c-Myc −/− mice are significantly smaller in size and lower in ploidy than those of control mice; however, because of the increase in megakaryocytic number and although fewer platelets are produced by each megakaryocyte, a greater-than-3-fold increase in platelet number was observed in Myc −/− mice. It is noteworthy that mice deficient in the RNA-binding motif protein 15 (Rbm15) gene (involved in human acute megakaryoblastic leukemia) develop a phenotype similar to that of Myc −/− mice: a lower number of LT-HSCs and a higher number of abnormal low-ploidy megakaryocytes. 55 Interestingly, the megakaryocyte number increase in Rbm15 knockout mice could be partially reversed by ectopic c-Myc. 55 The involvement of c-Myc in megakaryocytic differentiation is confirmed in transgenic mice with c-Myc overexpressed in the megakaryocytic lineage, attained with a Myc transgene under the control of the platelet factor 4 promoter. These mice show an increase in low-ploidy megakaryocytes owing to enhanced proliferation and survival, along with the blocking of differentiation. 56
Effects of Enforced Expression of c-Myc in Hematopoietic Cells In Vivo
Transgenic Mice Developing Lymphoid Neoplasia
The oncogenic function of c-Myc in hematopoietic malignancies was first demonstrated in the Eµ-Myc transgenic mouse, in which c-Myc expression is targeted to the lymphoid compartment by the immunoglobulin heavy chain gene promoter and enhancer. 57,58 Analysis of the immunoglobulin gene rearrangements of these tumor cells indicates that tumor development can commence at several points during B-lymphocyte differentiation, 17,57 suggesting that the deregulated Myc may be active at several developmental stages. The majority of the Eµ-Myc tumors appear after a latency period of 2 to 5 months and harbor mutations in the Arf-Mdm2-p53 pathway, 59,60 indicating the insufficiency of c-Myc to transform lymphoid cells by itself. Eµ-N-Myc mice also develop B-cell lymphoma. 61 An interesting model is that of mice that conditionally express a MYC transgene (repressed by tetracycline) in lymphoid cells because the tTA gene is under the Eµ control. 62 Most of the tumors induced in these mice are immature T-cell lymphoma. Interestingly, the tumors undergo a sustained regression upon inactivation of the transgene (i.e., adding tetracycline in the drinking water). 62
Although the Eµ-Myc-induced lymphoma demonstrates the ability of c-Myc to transform B cells in mice, the resulting tumors do not faithfully reproduce the Burkitt lymphoma, the hallmark of MYC-induced human B-cell lymphoma. Additional transgenic mice lines have been constructed to have better models for human Burkitt lymphoma. Yeast artificial chromosome (YAC) technology was used to obtain mice carrying a single copy of the 240-kb IgH/c-Myc translocation region. 63 B-cell tumorigenesis occurs in these translocus mice, even when the entire Eµ intron enhancer region is deleted. The phenotype of tumors from IgH/c-Myc YAC transgenic mice is reminiscent of B-cell acute lymphoblastic leukemia (B-ALL), 63 which is equivalent to Burkitt lymphoma in leukemic phase and represents about 5% of acute lymphoblastic leukemia (ALL). 64 Finally, a mouse transgenic was generated carrying a murine Myc cDNA inserted in the mouse IgH locus in a site that corresponds to the t(8;14) translocation break in Burkitt lymphoma or the t(12;15) in murine plasmacytoma. 65 These mice developed lymphoblastic B-cell lymphomas with a Burkitt-like morphology, diffuse large B-cell lymphomas and plasmacytomas (i.e., tumors of mature B cells). Thus, these mice model Burkitt lymphoma more precisely than do the Eµ-Myc and YAC-Myc mice, which do not develop plasmacytomas. 65
Signaling of the BCR cooperates with c-Myc in the genesis of B lymphomas. 66 Interestingly, tumors that grow in mice expressing an activated BCR differ from those found in Eµ-Myc mice and resemble Burkitt lymphoma. In contrast, BCR itself (in the absence of antigen stimulation) cooperates with c-Myc in tumorigenesis. The resulting tumors differ from those in the Eµ-Myc mice and those in Eµ-Myc/activated BCR and resemble a subset of chronic B-cell lymphocytic leukemia. 66 A parallel model to study c-Myc involvement in B-cell differentiation targets Myc to the IgH Cα locus in mice. In contrast to Eµ-Myc mice, Cα-Myc mice do not develop early B-cell lymphoma but show impaired primary and secondary humoral immune responses, failing to generate mature antibody-secreting plasma cells owing to increased apoptosis. 67 Only when deregulated c-Myc is combined with enforced expression of the antiapoptotic Bcl-xL gene do the mice develop plasmacytomas that reflect many features of human multiple myeloma. This transgenic model is the counterpart of mice carrying the t(12;15) translocation (IgH-Myc), which also develop plasmacitomas. 68
Transgenic Mice Developing Myeloid Neoplasia
Although there is less information on the impact of c-Myc on myeloid transformation, transgenic mice with Myc overexpression in the myeloid cells demonstrate the carcinogenic potential of c-Myc in the myeloid compartment. Mice carrying the human MYC proto-oncogene under the control of the promoter of murine GATA-1 promoter (an erythroid-specific gene) developed an early-onset, rapidly progressive erythroleukemia that resulted in death 30 to 50 days after birth. 69 More recently, Smith and coworkers generated several transgenic mice lines carrying the Myc gene under the control of the Vav promoter, which is active throughout all hematopoietic cell lineages (precursors and mature cells). Interestingly, the neoplasia lineage varied with the Myc expression level achieved in the transgenic mice. Aggressive T-cell lymphomas, as well as other hematopoietic abnormalities, predominated in the highest Myc-expressing transgenic mice, as expected from the broad expression of the transgene. 70 In contrast, in the lines expressing lower c-Myc levels, most tumors were late-onset monocytic tumors. 71 It is noteworthy that a 2-fold increase in c-Myc levels switched the phenotype from monocytic tumors to exclusively T-cell tumors. Thus, relatively low c-Myc levels can transform monocyte–macrophages but are insufficient to transform T lymphocytes. 71
Retroviral-Mediated Expression of MYC in Murine Hematopoietic Precursors
Several reports illustrated the leukemogenic effects of c-Myc in hematopoietic cells after the enforced expression in murine hematopoietic precursors via retroviral infection, although the neoplasia induction varies, depending on the virus, infected cells, and Myc gene. Bone marrow retrovirally transduced with Myc resulted in rapid development of acute myeloid leukemia (AML), 72 and fetal liver cells infected with a different retroviral vector resulted in long-latency lymphoma. 73 The infection of bone marrow progenitors in a p53−/− background also resulted in lymphoma. 74 Interestingly, upon culturing in vitro, a number of lymphoma-derived cells underwent myeloid differentiation. These cells switched from myeloid to lymphoid lineage and induced B-cell lymphomas when returning to in vivo conditions. 75 In another study, mice bone marrow cells transduced with N-Myc developed monoclonal and transplantable AML, but c-Myc retrovirus was not leukemogenic in the same system. 76 This model shows that N-Myc overexpression is highly oncogenic in mouse myeloid cells. In a recent report, bone marrow for lethally irradiated cells were repopulated with bone marrow cells expressing Myc, and the mice developed an AML-like disease. 77 The coexpression of several antiapoptotic genes of the BCL2 family accelerated leukemogenesis but did not change the myeloid phenotype of the leukemia. 77 Thus, in experimental models, c-Myc is able to induce lymphoid and myeloid neoplasia. The data agree with the observation that Myc is upregulated in radiation-induced AML in mice 78 and with the deregulation of MYC found in human leukemia, as discussed below.
Deregulation of MYC in Hematopoietic Cell Neoplasia
The original finding of translocations involving MYC in Burkitt lymphoma fueled intense research in other lymphoma and leukemia. MYC translocations have also been found at low frequencies in other lymphomas but not in lymphoblastic cell leukemia, except in the Burkitt leukemia variant (former acute lymphocytic leukemia L3). In addition, a significant fraction of MYC in Burkitt lymphoma carry mutations in the coding sequence, with several of them resulting in more stable mutant c-Myc proteins, thus increasing the c-Myc level. 79–81 However, no recurrent MYC translocations have been reported in other tumors, including other lymphoblastic or myeloblastic leukemia. 82 Table 2 shows a summary of MYC alteration in human leukemia and lymphoma.
Summary of Myc Alteration in Human Hematological Malignancies a

Myc in Human Lymphoid Leukemia
Lymphoid neoplasms can be broadly divided into those originating from immature lymphoid cells (T- and B-lymphoblastic leukemia and lymphoma) and the mature B- and T-cell neoplasms. The former include common leukemia, such as chronic lymphocytic leukemia (CLL), follicular lymphoma, diffuse large B-cell lymphoma, plasma cell myeloma (multiple myeloma), and Burkitt lymphoma, the first human tumor where MYC deregulation was identified. 83
ALL is a heterogeneous disease comprising different entities that show clonal expansion of leukemic lymphoblasts. 83,84 In human ALL, upregulation of c-Myc through different mechanisms has been reported. Translocations t(8;14), t(8;22), and t(2;8) involving MYC deregulation are present in 5% of adult ALL and 2% to 5% of children ALL. 84 Aberrant c-Myc stability has been reported in lymphoblastic leukemia cell lines and bone marrow samples from pediatric ALL patients. 85 Prolonged c-Myc protein half-life was not correlated with the MYC-stabilizing mutation, as found in Burkitt lymphomas. 85 The ETS family factor TEL2 has been shown to accelerate lymphoma development in Eµ-Myc transgenic mice, and interestingly, TEL2 and c-Myc expression levels were coordinately elevated in pediatric B-ALL patients, suggesting that both oncogenes cooperate in B lymphomagenesis. 86 Finally, more than 50% of human T-cell acute lymphoblastic leukemia (T-ALL) have activating mutations of NOTCH1, 87 and MYC is a direct transcriptional target of oncogenic Notch1. 88,89 Thus, it has been suggested that Notch1 mediates T-cell transformation at least in part by sustaining c-Myc levels (reviewed in References 90 and 91). Increased expression of MYC and c-Myc targets were recently found in T-ALL concomitant with LEF1 inactivation. 92
CLL is the most frequent leukemia in adults (almost 25% of all leukemia in the United States and Europe). 93,94 CLL is a slow-progression disease, with a median survival of 10 years, but it presents a marked variability: About one third of patients show a more aggressive form of the disease, with shorter survival periods. Interestingly, MYC mRNA expression is low in CLL, 95 a result confirmed at the protein level (JM Caraballo and J León, unpublished results). MYC expression is similar in the bad and good prognosis groups of CLL. In contrast, in the malignant lymphoma form of the disease, Richter syndrome, MYC expression increases. 96 Moreover, MYC translocation in CLL is associated with poor prognosis. 97
Rearrangements of the MYC oncogene are present in 15% to 50% of primary human multiple myeloma tumors, in many cases involved in complex rearrangements. 98,99 Frequent upregulation of MYC is also observed in plasma cell leukemia, a monoclonal gammopathy that can evolve from multiple myeloma. 100
c-Myc in Human Myeloid Leukemia
Myeloid neoplasms belong to 3 major groups: myeloproliferative neoplasms (including chronic myeloid leukemia [CML]), myelodysplastic syndromes, and AML. 101 Moreover, human AML is actually a heterogeneous group of neoplasias affecting the myeloid lineage. The former FAB (French-American-British) classification system of AML (M0 to M8 subtypes, attending to the differentiation type and stage) has been superseded by the World Health Organization classification, which identifies 15 diseases characterized by clinical presentation and recurrent chromosomal aberrations. 101 Understandably, most molecular studies to date are carried out with a relatively small number of cases of each myeloid disease or do not make distinctions among the different entities. Therefore, the information on MYC expression in myeloid leukemia (or expression of any gene, except those involved in recurrent translocations) refers to a rather heterogeneous group of diseases, and its possible involvement in a particular myeloid neoplasm has not been properly addressed.
The overexpression of MYC in bone marrow and peripheral blood in sporadic AML cases was observed early on. 102,103 Microarray-based studies and RT-qPCR studies showed that MYCN is over expressed in AML patients (as compared with normal bone marrow). 76 Recurrent translocations in AML generate fusion proteins that are leukemogenic transcription factors. 101 At least 3 of these—RUNX1-ETO, PML-RARα, and PLZF-RARα—induce c-Myc expression, suggesting that c-Myc is a downstream target of these oncogenes. 104,105
Amplifications of MYC in AML have been reported. 106–108 Double minute chromosomes and homogeneously staining regions containing amplified segments from chromosome band 8q24, including the MYC gene, have also been described. 106,109 However, it has been reported that a high MYC copy number does not result in higher c-Myc expression, 110,111 a situation that seems to conflict with the correlation between MYC amplification and expression observed in lymphoma. 112
MYC expression appeared elevated in a microarray-based study in 5 AML samples, as validated by RT-PCR. 113 In contrast, MYC was not detected as a major overexpressed gene in other microarray-based studies on AML samples. 114–116 Note, however, that microarray-based studies are done at the mRNA level; thus, changes in c-Myc protein level are not evaluated. Also note that MYC expression changes less than 2-fold are usually filtered out by the statistical analysis of microarray data but that a 2-fold expression change of c-Myc may be relevant. For instance, a mere 2-fold change means a major difference for c-Myc transformation ability of rat fibroblasts and mouse embryonic stem cells, 30,117 as well in transgenic animals where c-Myc dosage can be modulated. 118 It is noteworthy that in Burkitt lymphoma, the paradigm of MYC activation in human cancer, c-Myc increase in expression can be only 2-fold with respect to normal lymphocytes. 119 Finally, the great diversity of AML commented above makes the analysis of homogeneous sample cohorts difficult.
MYC mRNA is overexpressed in bone marrow cells from essential thrombocythemia, a myeloproliferative syndrome. 120 This is in contrast with the lack of MYC overexpression in another common myeloproliferative syndrome, the polycythemia vera (PV). 120 This is striking because more than 95% of PV carry activating mutation in the tyrosine kinase JAK2, which has been reported to induce MYC expression in cell lines, 121,122 suggesting that the pathway is not operative in PV primary cells.
CML is a common myeloproliferative disorder that progresses in 2 phases: Most patients are diagnosed in a relatively benign chronic phase, which is followed by an accelerated and, finally, blastic crisis phase. The molecular hallmark of all CML phases is the expression of the Bcr-Abl kinase, 123 which upregulates MYC expression 122,124 and cooperates with c-Myc in transformation. 125–127 Studies performed with a small number of cases showed that MYC mRNA levels were elevated in blastic crisis 128,129 and in chronic phase versus healthy bone marrow. 130,131 We recently observed the upregulation of c-Myc mRNA with CML progression (M Albajar et al., submitted). CML progression into blastic crisis is associated with cell survival, genomic instability, and differentiation arrest. 123,132 We have also shown that enforced MYC expression in CML-derived cells as K562 results in aberrant DNA synthesis under imatinib stress and block imatinib-mediated differentiation (M Albajar et al., submitted), suggesting that c-Myc may contribute to CML transformation. Note that trisomy 8 and gain at 8q24 (where MYC maps) are among the most frequent cytogenetic alterations in CML, 133,134 although their correlation with expression is unknown.
In conclusion, c-Myc function is pivotal for the correct hematopoiesis, helping to regulate the exquisite balance among self-renewal, differentiation, and proliferation required for blood formation. A reflection of c-Myc importance is the frequent finding of MYC deregulation in human leukemia and lymphoma, which would destroy this balance and transform hematopoietic cells by stimulating proliferation and blocking terminal differentiation.
Footnotes
Acknowledgements
We are grateful to Marta Albajar, Ana Batlle, and Ignacio Moreno de Alborán for a critical reading of parts of the article. We apologize to colleagues whose work was not cited in the form of their original articles but in reviews and whose work was not discussed because of space limitations or unintentional omission.
The work in our laboratories was supported by grants from the Ministerio de Ciencia e Innovacion of Spain (SAF08-01581) and the Red Temática de Investigación Cooperativa en Cancer (RD06/0020/0017) to J.L. and by a grant from Instituto Carlos III (FIS08/0829) to M.D.D.
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.