
Abstract
The MYC protein controls many cellular processes, including proliferation, cell cycle progression, cell growth, metabolism, angiogenesis, differentiation, cell adhesion, and motility. This is primarily achieved through transcriptional regulation of large gene networks that ultimately results in activation or repression of target genes. Given its broad regulatory scope, the expression of the MYC gene itself needs to be tightly controlled. Deregulation of MYC expression promotes tumorigenesis and, not surprisingly, MYC is frequently activated in many different human cancers. Furthermore, these tumors become highly dependent on sustained MYC expression, while MYC inactivation results in desirable anticancer effects, such as cell death, differentiation, and/or senescence. Thus, MYC has emerged as an attractive target for cancer therapy. In addition to regulating protein-coding genes, MYC also governs the expression of microRNAs, many of which have important regulatory roles in cancer development and progression. Here we will discuss how MYC-regulated miRNAs could be exploited for therapeutic development for cancer.
The Oncogenes of the MYC Family
Deregulated MYC activity is found in many human cancers and is predominantly the result of gene amplifications, translocations or point mutations of one of the MYC gene family members. Deregulation of MYC activity can also be achieved via upstream signaling pathways resulting in enhanced transcription, translation or increased stability of the MYC proteins (reviewed in References 1-5).
The MYC family genes (c-MYC, MYCN, MYCL) encode basic helix-loop-helix leucine zipper (bHLHZip) proteins, which, when dimerized with the small bHLHZip protein Max, bind to DNA at 5′-CACGTG-3′ motifs or similar E-box sequences. 6 Once bound to DNA, MYC/Max heterodimers recruit several additional cofactors necessary for gene regulation such as TRRAP (transformation/transcription domain-associated protein) and histone acetyl transferases (HATs). Thus, MYC-mediated transactivation of target genes occurs when MYC and Max are bound directly to E-boxes. 2,5-7 Further, as elegantly demonstrated in a recent genome-wide study, MYC-directed recruitment of the cofactors NELF and P-TEFb is required to support the release of promoter-proximal paused RNA polymerase II, 7,8 resulting in efficient transciptional elongation of MYC-bound genes. However, MYC also has the ability to repress target genes, when recruited by Miz-1 or other transcription factors to core promoters (see the reveiw by Herkert & Eilers 9 in this issue). Miz-1 itself acts as an activator of transcription. When the MYC-Max complex binds to Miz-1, the p300 HAT is displaced and the DNA methyl transferase DNMT3a is recruited. In this way MYC represses Miz-1 target genes. 6,10 Evidence that MYC also regulates gene expression via noncoding elements in the genome are accumulating rapidly. For example, the recent discovery that MYC regulates the expression multiple microRNAs 11,12 (miRNAs; reviewed by Bui & Mendell 13 in this issue) has intensified research to understand the biology associated with these findings.
The MYC network of transcriptional regulators also includes the Mad family, Mnt and Mga, all of which bind to the consensus E-box element as heterodimers with Max. 6,14 Transcriptional repression by Mad and Mnt is mediated through association with mSin3 that in turn recruits histone deacetylases (HDACs) and corepressors resulting in changes in chromatin structure. 15,16 Mnt-knockout mice die at birth whereas embryo fibroblasts from these mice show disrupted cell cycle control and can be transformed by Ras alone, traits resembling cells with MYC overexpression. 17 Further, Mnt is coexpressed with MYC in many proliferating cell types and has been suggested to be a modulator of MYC function. 14
MYC is an important regulator of many cellular processes, including proliferation, cell cycle progression, cell growth, metabolism, cell adhesion and motility, angiogenesis, and differentiation. 2,18,19 Further, when deregulated, MYC can induce immortalization, genomic instability, independence of growth factors, and escape from immune surveillance. 2,3 All of these functions promote growth and tumor formation, explaining why it is so often deregulated in cancer. Activated MYC can, however, also stimulate intrinsic tumor suppressor functions such as apoptosis and senescence. 18,20-24 The biological output of MYC activity has been shown to depend on the expression levels of MYC as well as the developmental context. 25-27
Why Is MYC an Attractive Target for Therapy?
Several biological features make MYC an attractive target for anticancer therapy. It is frequently activated in many human tumors, several of which are seemingly addicted to MYC. Addiction of tumors to MYC has perhaps been most clearly demonstrated in mouse models, where tumor regression has been observed upon MYC inactivation (reviewed by Felsher 28 in this issue). Reversibility of MYC-driven tumorigenesis was first shown in 2 different mouse models with conditional MYC expression. 29,30 In one of the studies, activation of MycER, a MYC-estrogen receptor fusion protein, in the suprabasal epidermis promoted papillomatosis accompanied by neoangiogenesis. In the other study, conditional expression of MYC in the hematopoietic system resulted in hematological malignancies. Importantly, in both cases deactivation of MYC led to spontaneous regression, demonstrating oncogene addiction of these tumors. 29,30 Tumor regression after switching off MYC expression has subsequently been observed in several mouse models with conditional MYC expression targeted to various tissues, including bone, pancreas, breast, and liver. 31-34 Depending on the malignancy studied, tumor regression was associated with apoptosis, cell cycle arrest, differentiation, and/or senescence. However, in several of these models, tumors reappeared when MYC was switched on again. 33-35 Remarkably, sustained regression after brief inactivation of MYC was reported in osteosarcoma and lymphoma models. 31,36
Concerns have, however, been raised regarding the feasibility of targeting MYC directly, as well as regarding the potential effects on rapidly regenerating tissues. Small molecules that can directly target MYC and inhibit its function have indeed been identified and characterized (reviewed by Prochownik & Vogt 37 in this issue). To address the second concern, Soucek et al. 38 engineered a transgenic mouse model carrying Omo-myc, which could be conditionally activated to inhibit the function of endogenous MYC. Omomyc is a mutant version of the bHLHZip domain of MYC designed to dimerize with wt MYC, and that also can dimerize with Max. 39 In addition, Omomyc has been shown to enhance MYC-induced apoptosis. 40 Induction of Omomyc in a Ras-driven lung cancer model with endogenous MYC expression efficiently induced regression, associated with apoptosis and senescence, of the tumors. 38 Furthermore, Omomyc expression had profound effects on normal regenerating tissues with high proliferative index, such as the hematopoietic system, skin, and intestines, but these effects were completely reversible and tolerated. Whether this holds true also in a human setting remains to be proven. Nevertheless, development of therapeutic strategies to selectively target MYC only in tumor cells will likely be of great importance. Another important issue that was not addressed in this study is what happens with the tumors after sequential deactivation of Omomyc expression. This is a fundamental question, which needs be tackled to truly model MYC inhibition as a future cancer therapy.
Strategies to Target MYC
Different approaches have been taken to find ways of therapeutically targeting MYC in cancer (reviewed in References 3, 4, 41). To date, strategies have aimed at either interfering with the tumor-promoting functions of MYC, such as proliferation and metabolism, or to stimulate tumor-suppressive functions induced by MYC, such as apoptosis and oncogene-induced senescence. 18 Approaches to directly target MYC include different ways of silencing MYC expression, or to modulate MYC protein stability. 3 Screening for small molecules that disrupt MYC/Max interaction is another strategy that has been successfully applied. 42-44 Several promising candidate compounds have been identified and are now further characterized and optimized with the aim for drug development (reviewed by Prochownik & Vogt 37 in this issue). Our own work has focused on cellular screening assays and has resulted in the identification of small molecules that induce apoptosis of tumor cells in a MYC-dependent manner. 45,46 Furthermore, our preliminary results, from a cellular screen using a collection of well-defined drugs and selected small molecules, indicate that only a subset of compounds showed a MYC-specific apoptosis response (Frenzel et al., manuscript in preparation). In a broader sense, drugs directed at upstream signal transduction pathways, which regulate MYC protein expression and stability or drugs acting on downstream MYC targets, are alternative ways to counteract MYC. For instance, the fact that MYC regulates multiple metabolic pathways has recently gained new attention, and the altered cancer metabolism is now being exploited for therapeutic development. 47,48 An alternative, and so far unexplored, route would be to tip the balance in the MYC network, i.e., to enhance the repressor functions of Mnt or Mad at the expense of MYC activity. In this review we will focus on the interplay between MYC and microRNAs and its potential use for therapeutic development in MYC-deregulated cancer.
Exploiting the MYC-miRNA Circuitry to Target Tumors
MicroRNAs are involved in the regulation of many biological processes, and their expression pattern is deregulated in cancer (reviewed in References 49-51 and Bui & Mendell 13 in this issue). A number of miRNAs have been shown to act either as oncogenes or as tumor suppressors and to be involved in initiation and progression of cancer. 51,52 Studies profiling miRNA expression have shown that the miRNA expression pattern can be used to classify tumors 53,54 and as a prognostic indicator. 55-58 Many studies have also implicated miRNAs as regulators of resistance to chemotherapy (reviewed in Reference 59).
A growing amount of work has shown that MYC regulates multiple miRNAs, many of which belong to miRNA families (reviewed by Bui & Mendell 13 in this issue). This includes both induction of miRNAs with oncogenic properties such as the miR-17~92 cluster 11,58,60-62 and repression of miRNAs with tumor suppressor function. 12 Interestingly, a broad repression of miRNA expression was seen upon overexpression of MYC. ChIP analysis showed that MYC was associated with the core promoters of the miRNAs it repressed. 12 The mechanism of the MYC-mediated repression was not addressed in this study. It was however partially resolved in a follow-up paper by Chang et al. 2009, 63 where the authors concluded that MYC-mediated transactivation of the RNA-binding protein Lin-28B is necessary for MYC’s ability to posttranscriptionally repress let-7 family members. However, these findings do not shed light on how MYC represses a large proportion of miRNAs directly at the promoter level. Suffice it to say, MYC-mediated repression of target genes (coding and noncoding) still requires clarification. In addition, MYC itself is regulated by miRNAs, some of which have documented tumor suppressor functions. 64,65
Little is known about miRNA regulation of the Mad family, Mnt and Mga. Only one publication has described Mnt as an important target of the hypoxia-induced miRNA miR-210. 66 Overexpression of miR-210, similar to knockdown of Mnt, triggered a MYC-like transcriptional response and promoted cell cycle progression. Antagonizing miR-210 could thus be an indirect way of counteracting MYC in hypoxic tumors.
miRNAs affect the expression of a network of target genes that often cooperate in the same biological process. Modulating one miRNA might therefore affect a pathway at multiple levels. Also, a single miRNA might regulate several oncogenic pathways. Many miRNAs that are regulated by MYC have well-established oncogenic or tumor suppressor functions and may be useful for anticancer therapeutic development irrespective of their targets (Fig. 1). miRNA-based therapies in cancers with deregulated MYC activity could consist of either inhibition of oncogenic miRNAs induced by MYC or reintroduction of miRNAs with tumor suppressor function that are repressed by MYC. Another possibility would be to use miRNAs that target MYC itself. Alternatively, miRNAs that regulate oncogenic pathways or tumor suppressor pathways that cooperate with or counteract MYC, respectively, could be exploited (Fig. 1). Here we will present some examples of how our growing knowledge about miRNA signaling can be used for potential therapeutic intervention in MYC-deregulated human cancers.
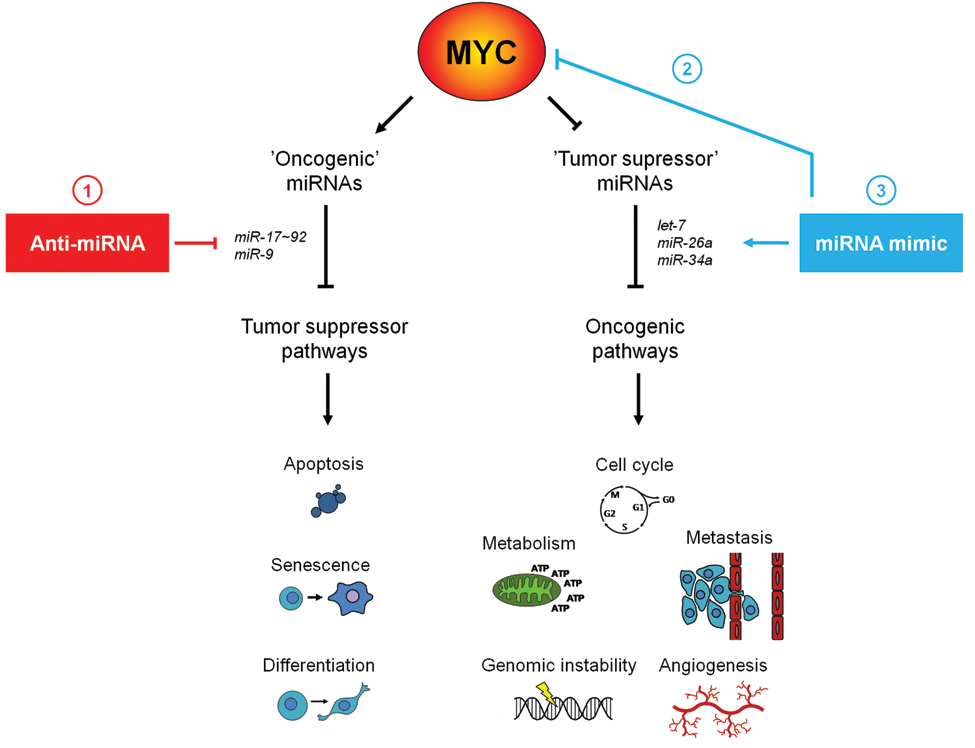
Strategies to target the MYC miRNA interplay in cancer. miRNA-based therapies could consist of either inhibition of oncogenic miRNAs using anti-miRNAs or of mimicking miRNAs with tumor suppressor functions. In cancers with deregulated MYC, anti-miRNAs could be directed against oncogenic miRNAs induced by MYC (
Counteracting Oncogenic miRNAs Induced by MYC
Oncomir-1, the miR-17~92 Cluster
Perhaps the most well-studied oncogenic miRNA cluster induced by MYC is the miR-17~92 polycistron, also known as oncomir-1. 67 This miRNA cluster is located within the noncoding gene C13orf25 at 13q31.3, which is frequently amplified in follicular lymphoma and diffuse large B cell lymphoma 68 and overexpressed in a variety of other tumors. Furthermore, oncomir-1 is a direct transcriptional target of MYC. 11 Six mature miRNAs—miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1—are encoded in this cluster. Although not tumorigenic in their own right when overexpressed in the lymphoid compartment, miR-17~92 cluster-derived miRNAs cooperate with c-MYC in inducing lymphomas in the Eµ-myc mouse lymphoma model. 62,69 Recently, miR-19 was identified as the key oncogenic component of the cluster in this model. In addition, miR-18 was also shown to have some oncogenic potential. 70-72 The tumor suppressor PTEN and the proapoptotic protein Bim have emerged as important targets repressed by oncomir-1–derived miRNAs in the hematopoietic system. 69-72
Further, miR-18a and miR-19a/b have been implicated in mediating the proangiogenic effect of MYC by targeting trombospondin-1 and connective tissue growth factor, thereby promoting tumor growth in vivo in a mouse colon carcinoma model. 73 MiR-92a, on the other hand, was shown to have an antiangiogenic effect when expressed in endothelial cells. 74 Moreover, miR-17 and miR-20a have been shown to regulate cell cycle progression by targeting E2F1. 11 Hence, the miRNAs in the miR-17~92 cluster regulate multiple and sometimes opposing functions involved in tumorigenesis.
In neuroblastoma, the miR-17~92 polycistron is a direct target of MYCN and subsequently overexpressed in tumors harboring amplification of the MYCN gene or with increased c-MYC/MYCN activity. 57,58,60-62 Neuroblastoma is a pediatric tumor originating from the developing sympathetic nervous system. Approximately 20%-30% of cases harbor amplification of the MYCN gene, which is associated with failure to respond to current therapies and therefore a very poor overall survival rate. 75 Several of the miRNAs from the oncomir-1 cluster have been shown to confer oncogenic properties to neuroblastoma cells. For example, miR-17 targets the proapoptotic BIM gene, as well as the p21 tumor suppressor gene (CDKN1A). Moreover, inhibition of miR-17 resulted in cell cycle arrest and apoptosis as well as inhibition of tumor growth of xenografted MYCN-amplified neuroblastoma cells. 61 Thus, repression of miR-17 can potentially inhibit neuroblastoma tumor growth in vivo.
Similarly, we have shown that antagonizing miR-18a and miR-19a in MYCN-amplified neuroblastoma cells resulted in cell cycle arrest. 62 Moreover, long-term miR-18a knockdown led to robust morphological and biochemical differentiation. This presumably involves the regulation of several genes, one of which was identified as estrogen receptor alpha (ESR1). Both miR-18 and miR-19 target ESR1, which, when overexpressed in MYCN-amplified neuroblastoma, promoted differentiation of the cells. 62 The degree of differentiation is especially important in neuroblastoma, since children with more differentiated tumors have a better clinical outcome than patients with less differentiated tumors. 76 Interestingly, a subset of neuroblastomas spontaneously regresses without any treatment. This is associated with a combination of differentiation and apoptosis of the tumor cells. 75,77 Mimicking this naturally occurring process has therefore been considered a plausible therapeutic avenue for neuroblastoma patients. Induction of differentiation could be used for residual neuroblastoma cells that are not killed by conventional cytotoxic drugs. The differentiating agent 13-cis-retinoic acid is used in this way in current therapeutic regimens in the treatment of minimal residual disease. 78,79 In summary, multiple members of the miR-17~92 cluster are excellent candidates for therapeutic development for MYCN-amplified neuroblastoma.
miR-9 and Metastasis
miR-9 is another miRNA with oncogenic properties that is induced by both c-MYC and MYCN 57,80 and that has been implicated in tumor cell invasiveness and metastasis. 80,81 One recent study showed that miR-9 promoted tumor cell invasiveness and metastatic potential in a breast cancer xenograft model. This was at least in part due to miR-9 mediated downregulation of E-cadherin, enabling the dissociation of carcinoma cells from one another and leading to release and activation of β-catenin and induction of VEGF expression. Conversely, inhibition of miR-9 decreased metastatic potential of highly metastatic tumors. 80,81 High expression levels of miR-9 correlated with the presence of metastasis in human breast cancer, and in hepatocellular carcinoma miR-9 was found to be the most upregulated miRNA in tumors with vascular invasion as compared to those without. 80,81 MYC overexpression and/or amplification have been associated with metastases and proposed to regulate metastasis formation in breast cancer in numerous studies. 82-86 Further, c-MYC activation is a common event in hepatocellular carcinoma and has been associated with more advanced disease and metastases. 87-89 Moreover, high miR-9 expression was found to correlate with the presence of MYCN amplification in neuroblastoma, which in turn is associated with metastatic disease and poor prognosis. 58,80 Taken together, these data support a role for miR-9 in mediating the invasive phenotype of MYC overexpression in various types of cancer
However, miRNAs can exert opposite biological activities depending on cellular context, as was recently demonstrated for miR-9 in 2 separate studies. 90,91 While miR-9 limits the migratory capacity of embryonic stem (ES)–derived human neural progenitor cells during early neural development, overexpression of miR-9 in adult mouse neural stem cells promoted their differentiation and migration. These seemingly opposing miR-9–mediated effects are attributed to the distinct repertoire of targets available to miR-9 during development and lineage expansion. Additional factors may also influence miR-9 function, including species-specific variations such as expression patterns and developmental stages. It remains to be determined in which context targeting miR-9 will be of benefit to cancer patients.
Restoring Expression of miRNAs that Are Repressed by MYC
miR-26a, Illustrating miRNA-based Therapeutic Targeting
Studies by Chang et al. 12 have shown that overexpression of MYC results in a broad repression of miRNA expression. Several of the miRNAs repressed by MYC, including let-7 miR-34a and miR-15a/16-1, have been shown to possess tumor suppressor function, indicating that reconstituting tumors with any of these miRNAs could be therapeutically beneficial in MYC-deregulated tumors. 55,56,92-100
In a recent study, Kota et al. 101 set out to prove the principle of systemic delivery of a single MYC-repressed miRNA as a therapeutic strategy for cancer. The authors reasoned that miRNAs that are highly expressed in normal tissue but reduced in tumor tissue would be well tolerated in normal tissue, but might exhibit antiproliferative or proapoptotic effects in the tumor. 101 miR-26 was chosen for these studies since it fulfilled the postulated criteria.
Using a MYC-driven mouse hepatocellular carcinoma model and an adenovirus associate virus (AAV) vector delivery system, it was shown that therapeutic reintroduction of miR-26a could reverse disease progression. Therapeutic delivery of miR-26a to established tumors in the liver led to tumor regression associated with apoptosis in the tumor cells. Further, the effect of miR-26a was tumor cell–specific since AVV-mediated delivery of this miRNA led to apoptosis induction in the cancer cells but not in nonmalignant hepatocytes. 101 Importantly, this study was the first to show that therapeutic delivery of a miRNA in a mouse model using a viral vector is feasible.
Introduction of miR-26a might also be beneficial for the human malignancy since miR-26a expression was found to be low in human liver cancer samples. Further, reintroduction of miR-26 into hepatocellular cancer cells resulted in reduced expression of cyclin D2 and cyclin E2 and induced cell cycle arrest. 101 This was further supported by the finding in a larger cohort of hepatocellular carcinoma patients, where low miR-26a expression was associated with shorter overall survival. 102 Although there are still many open questions regarding the importance and biological function of miR-26a in liver tumors, the importance of the study by Kota et al. 101 is the demonstration of the potential promise for therapeutic manipulation of miRNAs in cancer.
The let-7 Family of Tumor Suppressors
The let-7 family is another interesting group of miRNAs that are suppressed or deleted in various human cancers. 55,56,92,103 Several members of the let-7 family, which consists of 11 highly conserved miRNAs with identical seed sequence, have been found to be repressed by MYC. 12 A number of oncogenes, including Ras, MYC, and HMGA2 are targeted by let-7. 54,64,104-107 Numerous proteins associated with control of the cell cycle and proliferation are also regulated by let-7. 108
Reduced let-7 expression has been proposed to be a marker for less-differentiated tumors 54 and has been associated with poor prognosis in lung cancer. 55,56,109 In line with these findings, overexpression of let-7 in a lung cancer xenograft as well as in a Ras-induced mouse lung cancer model delayed tumor growth. 93,94 Importantly, therapeutic delivery of let-7 to established Ras-induced lung tumors in mice caused tumor regression. 110 let-7 replacement therapy therefore seems to be a promising modality to explore for the therapy of lung cancer.
miR-34a, Targeting MYCN in Neuroblastoma
Many miRNAs have been mapped to chromosomal regions that are frequently deleted in tumors. 92,99,111 Loss of heterozygosity of 1p36 was first identified in neuroblastoma, 112 where it correlates with MYCN amplification. 75 Later, loss of this chromosomal region was also observed in other human malignancies, suggesting that 1p36 harbors one or more tumor suppressor genes. Several genes located in this region have been proposed to act as tumor suppressor genes in neuroblastoma, but no consensus regarding a single key tumor suppressor gene has been reached. A number of miRNAs also map to this chromosomal region, among them miR-34a. 97 Reintroduction of miR-34a in 1p36del neuroblastoma cells or MYCN-amplified neuroblastoma cells with low levels of miR-34a lead to pronounced growth inhibition. 65,96,97 MYCN was shown to be a direct target of miR-34a. 65 Other proposed target genes include E2F3 and BCL2. 96,97 Hence, miR-34a regulates several oncogenic proteins, including MYCN. Furthermore, miR-34a is repressed directly by c-MYC, 12 suggesting that MYC family members have adopted various molecular strategies to avoid negative regulation by miR-34. Thus, in vivo analysis of the tumor suppressor function of this miRNA in a MYCN-driven neuroblastoma model would be interesting and important for further validation of miR-34a as a therapeutic strategy.
Therapeutic Targeting and Delivery of miRNAs in Cancer
The discovery of RNA interference has encouraged efforts into therapeutic development of siRNA-based interventions, with several ongoing clinical trials (reviewed in References 113, 114). Similar principles will apply to therapeutic delivery of miRNAs to tumors. Sufficient delivery of RNA to the tissue of interest seems to be a main obstacle. To date the greatest progress for RNA-based therapies has been in tissues where it can be applied locally, such as in the eye, and intranasal delivery to target the lungs. 113 Systemic delivery of RNA has proved to be more challenging, and many strategies for improvement are being evaluated.
There have been various approaches to modify RNA oligonucleotides to achieve increased stability and facilitated uptake into cells. Krützfeldt et al. 115 developed so-called antagomirs, which are cholesterol-linked 2′-OMe oligonucleotides targeting miRNAs. Treatment with antagomirs in mice led to repression of the target miRNA in all examined tissues except for the brain. Strikingly, one single injection could lead to silencing for as long as 23 days. 115 In addition, systemic administration of an antagomir targeting miR-10b resulted in reduced metastasis formation in a mouse breast cancer model. 116 Another approach with chemically modified anti-miRNAs have been to use locked nucleic acid–modified oligonucleotides (LNA-antimiRs), which displayed improved potency over 2′-OMe–modified oligonucleotides. Systemic delivery with an LNA-antimiR efficiently inhibited its target miRNA in mice as well as in African green monkeys, without evidence of LNA-associated toxicity or histopathological changes. 117 So far, no attempts to therapeutically target tumors with LNA-antimiRs in animal models have been reported.
Further, approaches using various delivery systems to achieve more efficient and specific delivery of RNAs are being explored. These include viral vectors, liposomes, and polymer-based nanoparticles, each with their own set of advantages and limitations (reviewed in References 113, 114). Polymer-based nanoparticles might be particularly promising since they allow the addition of targeting ligands to the surface of the nanoparticle. This approach would ensure the specific delivery of sufficient amounts of RNA to the tissue of interest. Targeting molecules can be ligands for cell type–specific receptors or antibodies directed at cell surface proteins expressed specifically by tumor cells. The first report on successful uptake of siRNA in tumors and downregulation of the target mRNA and protein after systemically administering the siRNA using a targeted nanoparticle delivery system in humans was recently published. 118 This opens up possibilities for using similar approaches for therapeutic delivery of miRNAs to human tumors.
Conclusion
Deregulation of MYC contributes to the development a wide variety of human tumors, including leukemia and neuroblastoma. Counteracting the tumor-promoting activity of MYC is therefore an appealing anticancer strategy. Approaches targeting different levels of the MYC pathway are currently being explored. With the characterization of the interplay between MYC and miRNAs, yet another layer of targeting opportunities has emerged. Thus the rapidly growing understanding of the biology of miRNAs in cancer as well as the development in RNA-mediated therapeutic approaches in recent years promises new, exciting modes of specific targeting in cancer.
Footnotes
Acknowledgements
We thank L.-G. Larsson for critical reading of this manuscript and all group members for fruitful discussions. Our research is supported by grants from the Swedish Research Council, the Swedish Cancer Society, the Swedish Childhood Cancer Foundation, the Hedlund Foundation, the King Gustaf V Jubilee Foundation, the Åke Olsson Foundation, and Karolinska Institutet. A.F. is supported by a postdoctoral position from the Swedish Childhood Cancer Foundation and J.L. by a postgraduate research grant from Karolinska Institutet. M.A.H. is a recipient of a Senior Investigator Award from the Swedish Cancer Society.
The authors declare no conflicts of interest with respect to the publication of this article.