
Abstract
Objective
This study aimed to evaluate the therapeutic potential of gymnoside I, a bioactive compound isolated from Bletilla striata, in the management of hemorrhage-related disorders and to elucidate its underlying mechanisms of action.
Methods
A multi-tiered experimental strategy was employed. In vivo assays assessed bleeding and clotting times in normal mice and examined gymnoside I’s effects on heparin-induced coagulopathy. Pharmacokinetic–pharmacodynamic (PK–PD) modeling was conducted to characterize time- and concentration-dependent effects. Network pharmacology was used to identify intersecting genes between gymnoside I and hemorrhagic diseases, followed by molecular docking and biolayer interferometry to validate protein interactions.
Results
Gymnoside I significantly reduced bleeding and clotting times in normal mice and partially reversed heparin-induced coagulopathy by improving thrombin time, platelet count, thromboxane B2, and fibrinogen levels. PK–PD modeling revealed distinct hysteresis loops, indicating delayed stimulation and potential feedback regulation. Network pharmacology identified 26 intersecting genes across purpura, cerebral hemorrhage, and aplastic anemia, with TNF, AKT1, and SRC emerging as key targets. Molecular docking predicted stable binding of gymnoside I to these proteins, and biolayer interferometry confirmed direct interactions, particularly with TNF.
Conclusion
Gymnoside I exerts multi-target modulation of coagulation and inflammatory pathways, supporting the concept of “treating different diseases with the same approach.” These findings provide mechanistic insight into its pharmacological activity and highlight its potential for further development in hemorrhagic disease therapy.
Keywords
1. Introduction
Hemorrhagic disorders, characterized by impaired coagulation and excessive bleeding, pose significant clinical challenges and are often associated with high morbidity and mortality.
1
These disorders encompass a wide spectrum of diseases, including purpura, cerebral hemorrhage, and aplastic anemia, which—despite differing in etiology and pathophysiology—share a common clinical manifestation of bleeding due to coagulation dysfunction or vascular fragility.
2
For instance, purpura involves capillary leakage and platelet abnormalities,
3
cerebral hemorrhage results from vascular rupture within the brain,
4
and aplastic anemia leads to pancytopenia, including reduced platelet counts that compromise hemostasis.
5
The convergence of bleeding symptoms across these distinct conditions highlights the need for therapeutic agents capable of addressing shared pathological mechanisms. Current therapeutic strategies, including anticoagulant reversal agents and procoagulant drugs, are limited by narrow mechanisms of action, potential side effects, and variable efficacy across different pathological contexts.
6
Moreover, the complexity of hemorrhagic conditions often requires multi-target interventions that can simultaneously regulate coagulation, inflammation, and vascular integrity. In this context, Traditional Chinese Medicine (TCM) offers a unique paradigm through the concept of yi bing tong zhi or “treating different diseases with the same therapeutic principle.”
7
This approach emphasizes the treatment of diverse diseases that share similar pathological features—such as bleeding—with a unified herbal strategy. Bletilla striata, a classical hemostatic herb in TCM, exemplifies this principle and has been widely used to treat various bleeding-related disorders.
8
Among its active constituents, gymnoside I has recently attracted attention for its potential multi-target pharmacological effects, and its chemical structure is shown in Figure 1.9,10 Chemical structure of gymnoside I
TCM offers a rich source of bioactive compounds with complex pharmacological profiles, many of which exhibit multi-target therapeutic effects. B. riata, a perennial orchidaceous herb, is one of the most prominent hemostatic agents in TCM. 8 Traditionally, it has been used to treat a variety of bleeding-related conditions, including hemoptysis, hematemesis, traumatic bleeding, ulcers, and tissue injuries. Its therapeutic applications are documented in classical texts such as the Compendium of Materia Medica, and modern clinical practice continues to validate its efficacy in managing hemorrhagic symptoms. 11 Recent pharmacological studies have increasingly focused on elucidating the molecular basis of B. striata’s hemostatic effects. Experimental evidence suggests that its bioactivity is not limited to physical astringency or wound coverage, but involves modulation of coagulation pathways, anti-inflammatory responses, and promotion of tissue regeneration. 12 In vivo and in vitro studies have demonstrated that extracts of B. striata can enhance platelet aggregation, shorten bleeding time, and reduce vascular permeability, indicating its potential in treating both internal and external bleeding disorders. Phytochemical investigations have identified a diverse array of constituents in B. striata, including polysaccharides, bibenzyl derivatives, phenanthrenes, and glycosides. 11 Among these, polysaccharides have been widely studied for their immunomodulatory and wound-healing properties. However, recent attention has shifted toward small-molecule compounds with more defined pharmacokinetic profiles and target specificity. gymnoside I, a bibenzyl glycoside isolated from B. striata, has emerged as a particularly promising candidate due to its unique structural features and preliminary pharmacological activity. 10 Early studies suggest that gymnoside I may exert regulatory effects on both coagulation and inflammation, two key pathological processes in hemorrhagic disorders. 10 Its potential mechanisms include modulation of thrombin activity, influence on platelet function, and interaction with inflammatory mediators such as TNF-α and IL-6. Moreover, its relatively low molecular weight and favorable solubility profile make it suitable for further pharmacokinetic and pharmacodynamic evaluation. Despite these promising findings, the precise molecular targets and signaling pathways involved in gymnoside I’s hemostatic action remain poorly understood, necessitating a more systematic and integrative investigation.
In the field of TCM research, the integration of computational and experimental methodologies has become increasingly essential for elucidating the complex pharmacological mechanisms underlying multi-component herbal formulations. Network pharmacology offers a systems-level approach to identify potential bioactive compounds, predict their molecular targets, and map the interactions between compounds, targets, and disease pathways. 13 This method aligns with the holistic philosophy of TCM and enables the visualization of multi-target therapeutic networks, thereby providing a theoretical framework for understanding the synergistic effects of herbal medicines. Molecular docking complements network pharmacology by simulating the binding interactions between small molecules and target proteins at the atomic level. It allows for the prediction of binding affinity and orientation, offering mechanistic insights into how specific compounds may exert their biological effects. 14 This structural perspective is critical for validating target predictions and guiding further experimental design. Biolayer interferometry (BLI), a label-free and real-time biosensing technology, provides direct experimental evidence of molecular interactions by quantifying the binding kinetics between compounds and target proteins. 15 BLI enhances the reliability of computational predictions by confirming physical interactions and determining parameters such as association rate, dissociation rate, and equilibrium dissociation constant. The combined application of network pharmacology, molecular docking, and BLI represents a powerful and comprehensive strategy in TCM research. It enables the transition from empirical observations to mechanism-based investigations, facilitating the identification of key active constituents, elucidation of multi-target mechanisms, and validation of compound–target interactions. This integrative approach not only strengthens the scientific foundation of TCM but also accelerates the discovery and development of novel therapeutic agents derived from traditional herbal sources. 16
To bridge the current knowledge gap, this study systematically investigates the pharmacological activity and underlying mechanisms of gymnoside I using a heparin-induced coagulation disorder model. A multi-dimensional strategy was adopted, encompassing in vivo efficacy evaluation, pharmacokinetic–pharmacodynamic (PK–PD) modeling, network pharmacology analysis, molecular docking, and BLI. This integrative approach enables a comprehensive assessment of gymnoside I’s therapeutic potential while uncovering mechanistic insights into its multi-target interactions. The findings not only highlight gymnoside I as a promising natural agent for the treatment of hemorrhagic disorders but also contribute to the evidence-based modernization of TCM.
2. Materials and Methods
2.1. Drug and Reagents
Gymnoside I (HY-N12127) was purchased from MedChemExpress (MCE, NJ, USA). Puerarin (110752-201816) was obtained from the National Institutes for Food and Drug Control (Beijing, China). All reference compounds used in this study had a purity greater than 98%. Sodium citrate (616C0520) was sourced from Solarbio Life Sciences (Beijing, China). Thrombin time (TT, 2300417) and plasma fibrinogen (FIB, 2300202) assay kits were purchased from Zhongqin Shidi Biotechnology Co., Ltd. (Taizhou, China). The TXB2 assay kit (20180910) was obtained from Jiancheng Bioengineering Institute (Nanjing, China). Recombinant proteins SRC (Ag22306), AKT (Ag26642), and TNF (Ag24020) were purchased from ProteinTech Group Co., Ltd. (Wuhan, China). PD MiniTrap G-25 desalting columns (28-9180-07) were sourced from Cytiva (Marlborough, MA, USA). Black 96-well microplates (902560) were obtained from LeSai Biotechnology Co., Ltd. (Jiangsu, China). Octet® SA biosensors (2412024011) were purchased from Sartorius Trading Co., Ltd. (Shanghai, China). LC–MS/MS-grade methanol, formic acid, and acetonitrile were obtained from Merck Co. Ltd. (Darmstadt, Germany). Distilled water was sourced from Watsons Group Co., Ltd. (Hong Kong, China).
2.2. Animals
Male SPF-grade Kunming mice (18–22 g) were purchased from Changsha Tianqin Biotechnology Co., Ltd. (License No. SYXK [Xiang] 2019-0014). All animals were housed separately under standardized conditions in a temperature- and humidity-controlled environment (24 ± 2°C, 45%–75% humidity) with free access to food and water. Following a 7-day acclimatization period, experimental procedures were initiated.
2.3. Solution Preparation
Puerarin was accurately weighed and dissolved in methanol in a 10 mL volumetric flask to prepare a stock solution at a concentration of 0.495 mg/mL. This stock solution was subsequently diluted with methanol to obtain internal standard working solutions at a concentration of 20 ng/mL. All prepared solutions were stored at −20 °C until analysis. Similarly, gymnoside I was precisely weighed and dissolved in methanol to yield a stock solution at a concentration of 0.500 mg/mL. For the rat pharmacokinetic study, the standard solution was spiked into blank plasma to prepare calibration standards and quality control (QC) samples.
2.4. Sample Preparation
To prepare plasma samples, 100 µL of plasma was combined with 50 µL of internal standard (20 ng/mL puerarin), 50 µL of 2% formic acid, and 400 µL of methanol. After vortexing for 1 minute, the mixture underwent 10 minutes of sonication, followed by centrifugation at 12,000 rpm for 10 minutes at 4 °C. The supernatant was then dried under a nitrogen stream at 37 °C, reconstituted in 150 µL of 50% methanol, and subjected to a second centrifugation under identical conditions. The resulting solution was collected for subsequent UPLC–MS/MS analysis.
2.5. UPLC-MS/MS Conditions
Quantitative analysis of pharmacokinetic samples was performed using an ACQUITY™ UPLC system (Waters, Milford, MA, USA) equipped with a BEH C18 column (100 × 2.1 mm, 1.7 μm). The mobile phase consisted of water (A) and acetonitrile (B), each containing 0.2% formic acid, delivered at a flow rate of 0.30 mL/min. The gradient elution program was as follows: 0–1.5 min, 10% B; 1.5–4.0 min, linear increase from 10% to 90% B; 4.0–6.5 min, 90% B; and 6.5–7.0 min, re-equilibration from 90% to 10% B.
Mass Spectrometric Conditions of Gymnoside I and IS
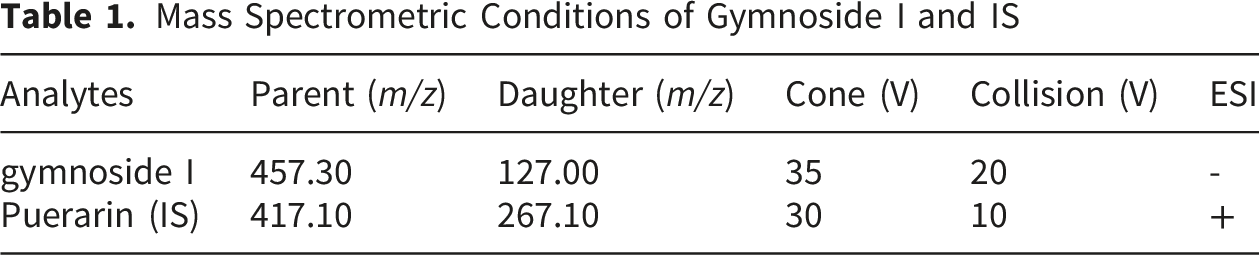
2.6. Effects of Gymnoside I on Bleeding Time and Clotting Time
Fifty SPF-grade Kunming mice were randomly assigned to five groups: a blank control group (CON), a positive control group (POS) treated with the hemostatic agent etamsylate (Eta; 40 mg/kg), and three gymnoside I treatment groups receiving low (Gym I-L; 10 mg/kg), medium (Gym I-M; 20 mg/kg), and high (Gym I-H; 40 mg/kg) doses. Each group received daily tail vein injections of the respective treatments for five consecutive days. The CON group was administered normal saline (10 mL/kg) as a vehicle control. On day 5, sixty min after the final administration, CT was measured using the capillary tube method. A glass capillary tube (diameter: 1 mm; length: 100 mm) was inserted into the retro-orbital venous plexus, and timing began once blood entered the tube. After the tube was filled, it was placed horizontally on a flat surface. Every 10 s, the tube was broken at 0.5 cm intervals and gently pulled apart to observe the formation of a fibrin thread. The time elapsed until the appearance of the fibrin thread was recorded as the CT. BT was assessed by tail transection. Approximately 3 mm of the tail tip was removed, and timing began as blood started to flow. Every 10 s, the wound was gently blotted with filter paper until bleeding ceased. The total time from the onset of bleeding to cessation was recorded as the BT.
2.7. Effects of Gymnoside I on Coagulation Function in Mice
Sixty SPF-grade Kunming mice were randomly divided into six groups: a blank control group (CON), a heparin-induced model control group (MOD), a protamine sulfate group (POS; 1.6 mg/kg), and three gymnoside I treatment groups. All mice re-ceived daily oral gavage of the respective treatments for five consecutive days. The CON group was administered normal saline (10 mL/kg), while the other groups re-ceived their designated compounds at the specified doses. On day 5, all groups except CON were injected with heparin sodium (0.8 U/mouse) via the tail vein to induce a coagulation disorder model; the CON group received an equivalent volume of normal saline. Sixty minutes after the final oral administration, blood was collected via enucleation. PL was measured immediately. Blood samples were transferred into EP tubes pre-coated with sodium citrate and centrifuged at 6000 r/min for 10 minutes to obtain plasma. The plasma was then used for the determination of TXB2, TT, and FIB levels.
2.8. PK-PD Model
Ten Kunming mice were subjected to the same modeling and dosing protocol as the Gym I-H group outlined in Section 4.7. Tail vein blood samples (0.25 mL) were collected before administration and at 0.17, 0.33, 0.5, 1, 1.5, 2, 4, 8, 12, 24, 36, and 48 h following the final dose. To ensure fluid homeostasis, each blood draw was followed by an equal-volume injection of normal saline. Plasma was isolated via centrifugation at 6000 r/min for 10 min at 4 °C, and 200 μL aliquots were stored at −80 °C for PK and PD evaluation.
Quantitative analysis of gymnoside I plasma concentrations, along with pharmacodynamic biomarkers including PT, TXB2, TT, and FIB, was conducted to characterize the PK–PD relationships. Model selection was based on Akaike’s Information Criterion (AIC) to ensure optimal fit and parsimony. The pharmacokinetic profile of gymnoside I in heparin-induced rats was best described by a two-compartment model. For the pharmacodynamic component, the Sigmoid Imax or Sigmoid Emax models provided the best performance in capturing the concentration–effect relationships, accurately reflecting the nonlinear dynamics between drug exposure and biomarker response:

2.9. Target Prediction and Network Analysis
Potential targets of gymnoside I were predicted using multiple databases, including TargetNet (https://targetnet.scbdd.com/), BATMAN-TCM (https://bionet.ncpsb.org.cn/batman-tcm/), and SwissTargetPrediction (https://www.swisstargetprediction.ch/), with species restricted to Homo sapiens. Disease-related target genes associated with “purpura,” “cerebral hemorrhage,” and “aplastic anemia” were retrieved from the DrugBank (https://www.drugbank.ca), Therapeutic Target Database (https://db.idrblab.net/ttd/), OMIM (https://www.omim.org/), DisGeNet (score > 0.1, https://www.disgenet.org/search), and GeneCards (score > 10, https://www.genecards.org/), also limited to human targets. All target names, IDs, and species were verified using UniProt (https://www.uniprot.org/). The results from each database were imported into Excel, and duplicate disease-related genes were removed. A union of the non-redundant gene sets was compiled to represent the full set of disease-associated targets. Venn diagram analysis was then performed to identify overlapping targets among gymnoside I and the three diseases, which were considered as potential “shared therapeutic targets” for hemorrhagic conditions. These intersecting genes were imported into R software for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Protein–protein interaction (PPI) data were retrieved from the STRING database (https://cn.string-db.org/), with species restricted to Homo sapiens. The resulting network was analyzed using the NetworkAnalyzer tool in Cytoscape 3.7.2 (https://cytoscape.org/) to assess topological features and identify key therapeutic targets. The top three targets ranked by degree value were selected for subsequent molecular docking and BLI validation experiments.
2.10. Molecular Docking Simulation
The chemical structure and PubChem ID of gymnoside I were retrieved from PubChem (https://pubchem.ncbi.nml.gov). Ligand files were converted from SMILES format to PDBQT using AutoDockTools version 1.5.6 (The Scripps Research Institute, CA, USA), with appropriate ligand preparation settings applied prior to docking. Crystallographic structures or homology models of key target proteins (AKT1, TNF, and SRC) were obtained from the Protein Data Bank (PDB, https://www.rcsb.org/pdb). Receptor proteins were preprocessed using AutoDockTools by removing water molecules and native ligands, followed by hydrogenation. The docking grid box was defined by specifying the center coordinates and setting the number of grid points along the X, Y, and Z axes. Protein–ligand interactions were analyzed using the Protein–Ligand Interaction Profiler and visualized with PyMOL. A binding energy threshold of less than −5 kcal/mol was considered indicative of significant binding affinity.
2.11. Biolayer Interferometry
To assess the binding affinity between gymnoside I and recombinant proteins AKT1, TNF, and SRC, each protein was dissolved in 5% trehalose in PBS at 200 μg/mL, gently mixed, aliquoted, and stored at −80 °C. Proteins were biotinylated using a freshly prepared 10 mM biotin solution in DMSO, with reagent volumes calculated based on protein properties. After 45 min of incubation at room temperature, biotinylated proteins were purified using PBS-equilibrated desalting columns and stored at 4 °C. Streptavidin biosensors were pre-wetted in PBS and used to immobilize the biotinylated proteins in a 96-well plate. Immobilization was performed using BLI with the following steps: Baseline (60 s), Loading (800 s), and Baseline (60 s), with loading time adjusted based on signal strength.
Gymnoside I was dissolved in PBS with 3% DMSO to prepare a 1 mg/mL working solution. Serial dilutions were made to create six concentration gradients, using matching buffer for dilution and blank controls. Binding assays were conducted using Octet BLI Discovery software (v13.0, Sartorius AG, Germany), following a Baseline–Association–Dissociation protocol (60 s each). Data were analyzed with Octet Analysis Studio (v13.0, Sartorius AG, Germany), applying double referencing to generate binding curves for gymnoside I with each protein.
2.12. Data Analysis
Data were presented as mean ± standard deviation (SD). PK-PD parameters were analyzed using WinNonlin software version 8.2 (Pharsight Corporation, Mountain View, CA, USA). Statistical analyses were conducted using SPSS version 23.0 (IBM Corp., Chicago, IL, USA). One-way analysis of variance (ANOVA) was applied for comparisons among groups, and a P value < 0.05 was considered statistically significant.
3. Results
3.1. Effects of Gymnoside I on BT and CT in Normal Mice
As shown in Figure 2, gymnoside I significantly reduced bleeding time (A, BT) and clotting time (B, CT) in mice in a dose-dependent manner following 7 consecutive days of tail vein injection. Compared to the control group, all tested doses of gymnoside I, as well as the positive control drug, produced statistically significant hemostatic effects (P < 0.05 or P < 0.01). Effect of gymnoside I on BT (A) and CT (B) in normal mice. Data were presented as Mean ± SD (n = 10); *P<0.05, **P<0.01 compared with the normal group
3.2. Effect of Gymnoside I on Coagulation Function in Systemically Heparinized Mice
As shown in Figure 3, systemic administration of heparin sodium significantly prolonged plasma thrombin time (TT) in mice (P<0.01), while levels of thromboxane B2 (TXB2), platelet count (PL), and fibrinogen (FIB) were markedly reduced (P<0.01), indicating impaired coagulation function. Following intervention with protamine sulfate and various doses of gymnoside I, TT was significantly shortened in all treatment groups (P<0.05), and TXB2, PL, and FIB levels were notably increased (P<0.05 or P<0.01). These findings suggest that gymnoside I may exert procoagulant effects by modulating the coagulation cascade and influencing key factors within the platelet system. Effects of gymnoside I on coagulation and platelet function in heparinized mice. Data were presented as Mean ± SD (n = 10); *P<0.05, **P<0.01 compared with the normal group; ##P<0.01 compared with the model group
3.3. Method Validation for Gymnoside I Quantification in Plasma
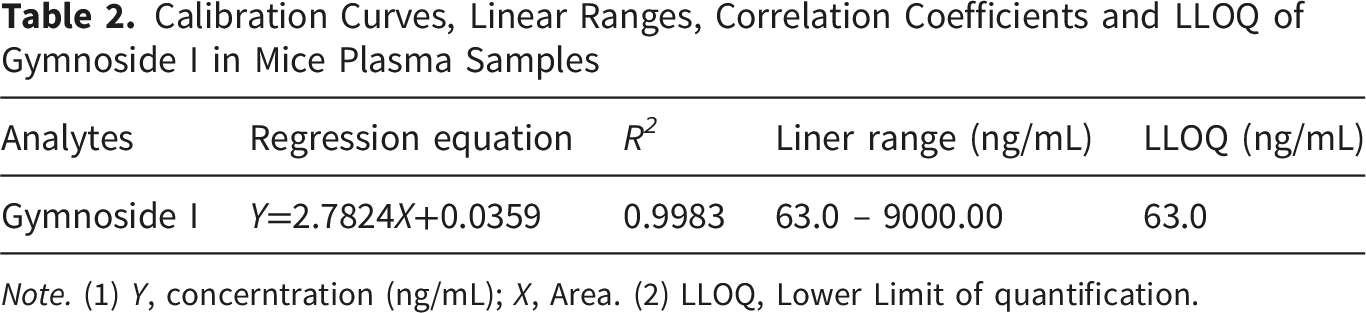
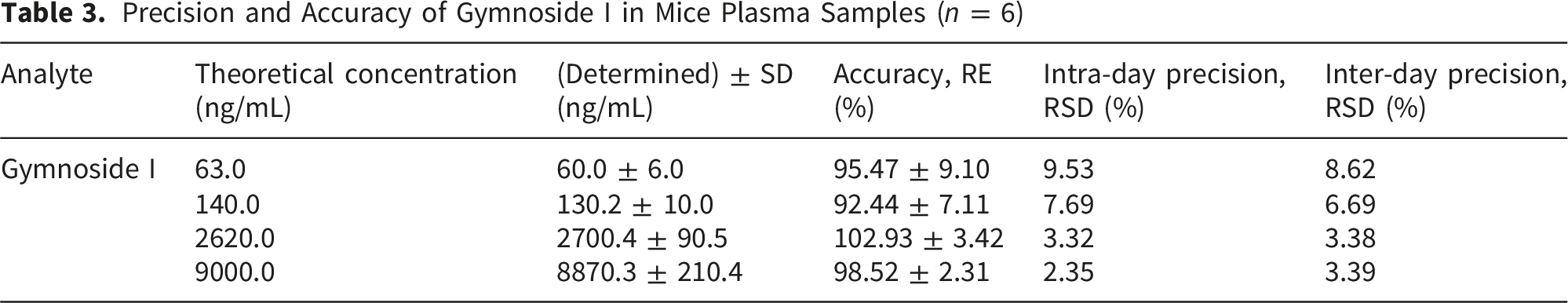
The UPLC–MS/MS method for determining gymnoside I concentrations in plasma samples was validated in accordance with Chinese Pharmacopoeia standards, covering key parameters such as specificity, linearity, sensitivity (LLOQ), accuracy, precision, recovery, matrix effects, and stability. Specificity was verified by comparing chromatograms of blank plasma, plasma spiked with gymnoside I at the LLOQ and puerarin, and actual sample extracts, confirming the absence of interference from endogenous components (Figure 4). The calibration curve exhibited excellent linearity (R2 > 0.99), with a lower limit of quantification established at 63.0 ng/mL (Table 2). Both intra- and inter-day assessments showed acceptable precision (RSD < 15%) and accuracy (RE within ±15%). Recovery ranged from 89.33% to 94.13%, and matrix effects were minimal (88.03–104.3%), as shown in Tables 3 and 4. Stability evaluations under various conditions (room temperature, 4°C for 24 h, and repeated freeze–thaw cycles) demonstrated consistent analyte integrity, with RSD values remaining within ±15% (Table 5). Typical UPLC-MS/MS chromatograms of blank plasma samples (A), blank plasma samples spiked with gymnoside I at LLOQ and puerarin (B), and actual plasma samples (C) Calibration Curves, Linear Ranges, Correlation Coefficients and LLOQ of Gymnoside I in Mice Plasma Samples Note. (1) Y, concerntration (ng/mL); X, Area. (2) LLOQ, Lower Limit of quantification. Precision and Accuracy of Gymnoside I in Mice Plasma Samples (n = 6) Recovery and Matrix Effect of Gymnoside I in Mice Plasma Samples (n = 6) Results of Sample Stability in Plasma Samples Under Different Conditions (n = 6)


3.4. PK–PD Analysis of Gymnoside I in Mice
The PK–PD analysis revealed that gymnoside I exerts time- and concentration-dependent effects on key coagulation biomarkers, including TXB2, TT, PL, and FIB (Figure 5A). Following administration in systemically heparinized mice, gymnoside I rapidly reached peak plasma concentrations and gradually declined, accompanied by dynamic changes in the pharmacodynamic markers. TXB2 and FIB levels increased in parallel with gymnoside I concentration, indicating a stimulatory effect on platelet activation and fibrinogen synthesis. In contrast, TT values decreased as gymnoside I levels rose, suggesting enhanced thrombin activity. PL also showed a positive correlation with gymnoside I exposure, further supporting its procoagulant potential. These findings demonstrate that the pharmacological activity of gymnoside I is closely linked to its systemic concentration, exerting a coordinated influence on coagulation pathways. The concentration-time-effect curves (A) and concentration-effect curves (B) of gymnoside I with TXB2, TT, PL, and FIB in the plasma of heparin-induced mice
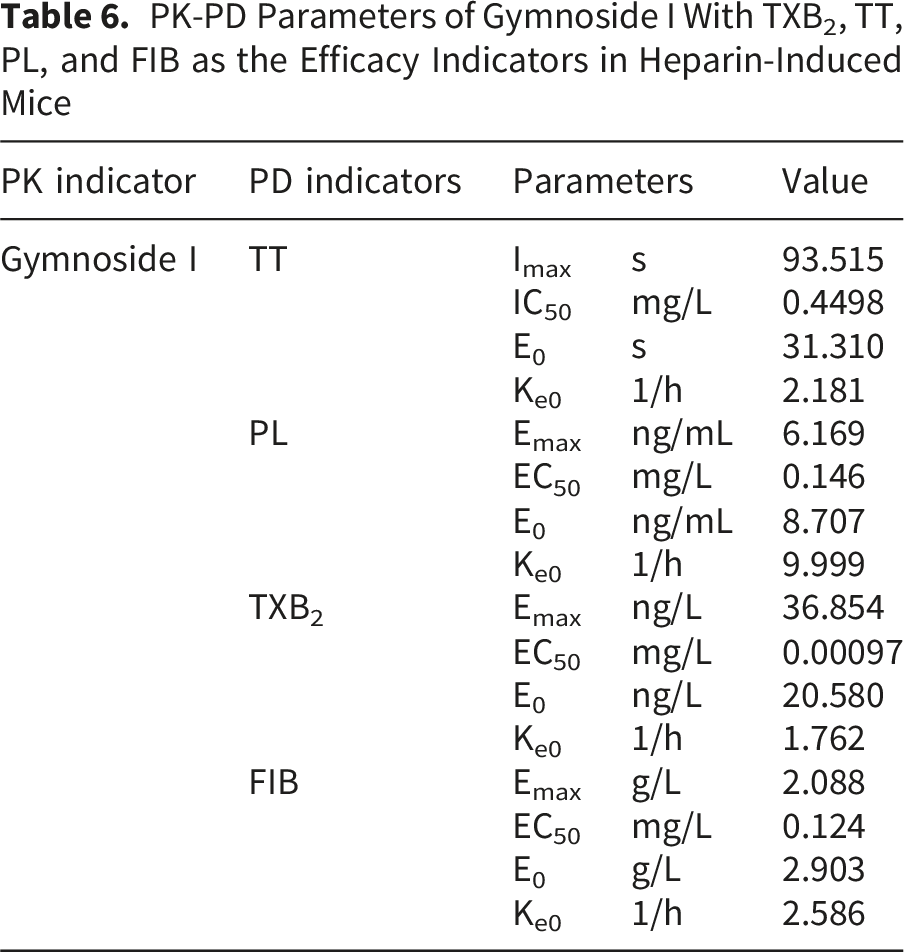
Effect–concentration curves were generated using a semi-compartmental modeling approach (Figure 5B). In PK–PD correlation analysis, the shape of the hysteresis loop provides insight into the temporal relationship between drug concentration and effect. A counterclockwise hysteresis loop typically indicates a delayed onset of effect at a given concentration, often due to the presence of an effect compartment or indirect pharmacological actions, such as those mediated by active metabolites. Conversely, a clockwise hysteresis loop suggests a reduction in drug effect over time at a constant concentration, commonly attributed to acute tolerance or the formation of inhibitory metabolites. In this study, the effect–concentration curves revealed that gymnoside I exhibited a counterclockwise hysteresis loop relative to TXB2, implying a delayed stimulatory effect likely mediated by downstream signaling or metabolite activity. In contrast, a clockwise hysteresis loop was observed relative to PL, suggesting the development of acute tolerance or feedback inhibition in platelet activation pathways. These distinct hysteresis patterns underscore the complex and multifaceted pharmacodynamic behavior of gymnoside I in modulating coagulation-related biomarkers.
PK-PD Parameters of Gymnoside I With TXB2, TT, PL, and FIB as the Efficacy Indicators in Heparin-Induced Mice
3.5. Network Pharmacology Results
To elucidate the potential therapeutic mechanisms of gymnoside I in hemorrhagic conditions, a comprehensive network pharmacology approach was employed. As shown in Figure 6A, a Venn diagram analysis was conducted to identify overlapping targets among gymnoside I and three hemorrhage-related diseases: purpura, cerebral hemorrhage, and aplastic anemia. A total of 26 intersecting genes were identified, suggesting these may serve as shared therapeutic targets relevant to the pathophysiology of hemorrhagic disorders. Figure 6B illustrates the protein–protein interaction (PPI) network constructed from these 26 intersecting genes using STRING database data. Each node represents a gene, and edges denote known or predicted interactions. The network reveals a high degree of connectivity, indicating that these genes may participate in coordinated biological processes such as platelet activation, immune modulation, and vascular integrity. To further assess the topological importance of individual genes within the network, degree centrality analysis was performed using Cytoscape, as shown in Figure 6C. Genes with the highest degree values (TNF, AKT1, SRC) were considered key regulatory nodes and were prioritized for subsequent molecular docking and BLI validation, given their potential role in mediating gymnoside I’s pharmacological effects. Functional enrichment analyses were conducted to explore the biological relevance of the intersecting genes. Figure 6D presents the GO enrichment results, highlighting significant terms across three categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The enriched GO terms suggest involvement in platelet activation, extracellular matrix organization, and signal transduction. Figure 6E displays the KEGG pathway enrichment analysis, where key pathways such as platelet activation, complement and coagulation cascades, and hematopoietic cell lineage were significantly enriched. These findings underscore the potential of gymnoside I to modulate critical pathways implicated in hemorrhagic disease mechanisms. Collectively, these results provide a systems-level perspective on the molecular targets and pathways through which gymnoside I may exert therapeutic effects, offering valuable insights for future experimental validation and drug development strategies. Network pharmacology analysis of gymnoside I and its potential therapeutic targets in hemorrhagic diseases. (A) Venn diagram showing 26 shared targets between gymnoside I and hemorrhagic disease-related genes. (B) PPI network of intersecting genes constructed via STRING. (C) Top hub genes identified by degree analysis in Cytoscape. (D) GO enrichment analysis of intersecting genes across BP, CC, and MF categories. (E) KEGG pathway analysis highlighting key signaling pathways
3.6. Molecular Docking Results
To elucidate the potential molecular mechanisms underlying the hemostatic and anti-inflammatory effects of gymnoside I, molecular docking simulations were performed against three key protein targets: AKT1, SRC, and TNF, which were identified by network pharmacology analysis as the top three genes with the highest degree values and considered key regulatory nodes. The docking studies were conducted using AutoDockTools, and the resulting complexes were visualized with PyMOL. Gymnoside I demonstrated favorable binding affinities to all three targets, with calculated binding energies below −5 kcal/mol, indicating strong and stable interactions.
In the TNF-Gymnoside I complex (Figure 7A), the ligand exhibited strong binding across multiple chains of the TNF trimer. Hydrogen bonds were formed with TYR-119, LEU-120, GLY-121, GLN-125, and TYR-151, with dual hydrogen bonding observed at GLY-121 and TYR-151. Hydrophobic interactions with LEU-57, TYR-59, and TYR-119 were also present, particularly near the receptor-binding interface. These interactions imply that gymnoside I may competitively inhibit TNF-receptor binding, thereby mitigating pro-inflammatory signaling cascades. In the AKT1-Gymnoside I complex (Figure 7B), the ligand was found to engage in a diverse array of noncovalent interactions within the kinase domain. Hydrogen bonds were formed with residues ARG-273, LYS-307, CYS-310, ASN-279, GLU-278, and THR-291, while salt bridges were observed with ARG-273 and LYS-297. Hydrophobic contacts involving LEU-156, VAL-164, THR-211, THR-291, and ASP-292 further stabilized the complex. Additionally, a π-stacking interaction with PHE-161 was identified, suggesting aromatic alignment near the ATP-binding pocket. These interactions collectively support the hypothesis that gymnoside I may modulate AKT1 activity, thereby influencing platelet activation and vascular signaling. Docking with SRC revealed similarly robust interactions (Figure 7C). Gymnoside I formed hydrogen bonds with ARG-178, SER-180, GLU-181, THR-182, and ILE-217, and salt bridges with ARG-158 and ARG-208. Hydrophobic interactions were noted with TYR-205, LEU-206, and ILE-217, located near the kinase domain. These findings suggest that gymnoside I may interfere with SRC’s catalytic function, potentially attenuating tyrosine phosphorylation events involved in platelet aggregation and endothelial permeability. Taken together, the docking results suggest that gymnoside I exerts its pharmacological effects through multi-target engagement, involving AKT1, SRC, and TNF. The presence of multiple stabilizing interactions—including hydrogen bonds, salt bridges, hydrophobic contacts, and π-stacking—across all three targets supports its potential as a therapeutic agent for hemorrhagic and inflammatory conditions. Docking poses of gymnoside I with TNF (A), AKT1 (B), and SRC (C). Key hydrogen bonds, salt bridges, and hydrophobic interactions are highlighted. All complexes show binding energies below −5 kcal/mol, indicating favorable binding affinity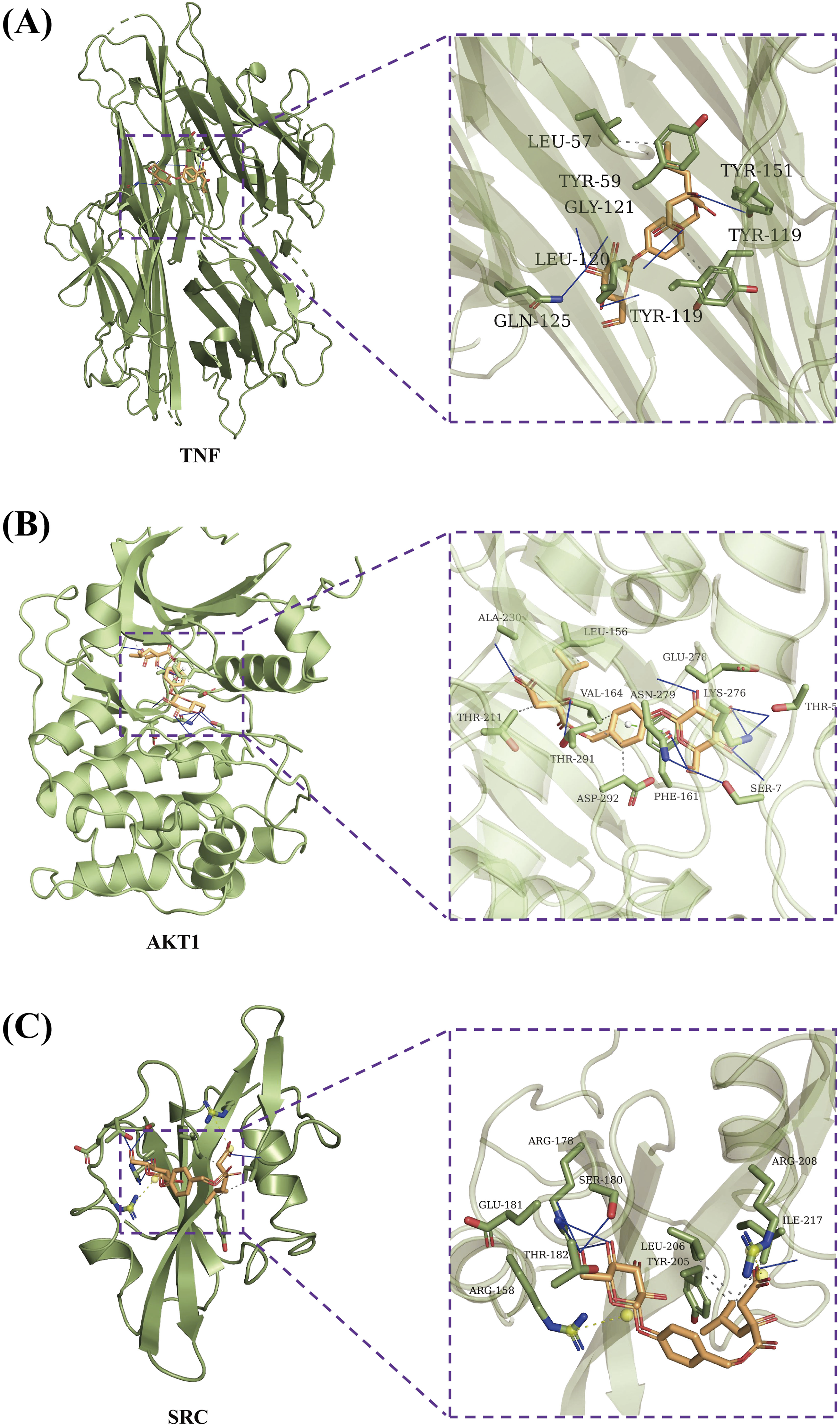
3.7. BLI Analysis
BLI was employed to assess the binding interactions between gymnoside I and the recombinant proteins TNF, AKT1, and SRC. Biotinylated proteins were immobilized on streptavidin biosensors, and gymnoside I was tested across a series of concentrations to determine binding kinetics and affinity. Figure 8A illustrates the binding profile of gymnoside I with TNF. The response signals increased proportionally with concentration, yielding a dissociation constant (KD) of 87.6 µM. This indicates a strong binding affinity, further supported by the sensorgrams showing distinct association and dissociation phases. Figure 8B shows the interaction between gymnoside I and AKT1, with a KD of 181.4 µM. Although slightly weaker than TNF, the binding remains robust and concentration-dependent, confirming a direct and specific interaction. Figure 8C presents the binding of gymnoside I to SRC, with a KD of 174.0 µM. The response curves demonstrate consistent and reproducible binding behavior, comparable to that observed with AKT1. Overall, gymnoside I exhibits strong binding affinity toward all three target proteins, with the highest affinity observed for TNF. These results support the potential of gymnoside I as a multi-target compound capable of modulating key signaling pathways involved in inflammation and cellular regulation. Biolayer interferometry analysis of gymnoside I binding to TNF, AKT1, and SRC proteins. BLI sensorgrams showing gymnoside I binding to TNF (A), AKT1 (B), and SRC (C). The compound binds all three proteins in a concentration-dependent manner, with KD values of 87.6 µM (TNF), 181.4 µM (AKT1), and 174.0 µM (SRC)
4. Discussion
B. striata, commonly known as Baiji, is a traditional Chinese medicinal herb renowned for its hemostatic, anti-inflammatory, and wound-healing properties. Historically, it has been extensively employed in the treatment of bleeding disorders, peptic ulcers, and traumatic injuries.17,18 In recent years, interest has focused on its diverse bioactive constituents, among which gymnoside I has shown promising preclinical activity. Evidence indicates that gymnoside I modulates coagulation cascades and inflammatory responses, supporting its efficacy in hemorrhage-related conditions. 19 With multi-target interactions and a favorable pharmacokinetic profile, gymnoside I is a strong candidate for drug development. Its systems-level activity also reflects the TCM principle of “treating different diseases with the same approach”. 20 This study presents a comprehensive pharmacological evaluation of gymnoside I, integrating in vivo assays, PK–PD modeling, network pharmacology, molecular docking, and biolayer interferometry. Through this multi-dimensional approach, the therapeutic potential of gymnoside I in hemorrhagic disorders is systematically elucidated. The findings not only confirm its efficacy in regulating key coagulation parameters but also reveal its multi-target engagement and integrative biological activity. These insights provide a mechanistic foundation for the traditional use of B. striata and support the rational incorporation of gymnoside I into modern therapeutic strategies for hemorrhage-related diseases.
One of the most compelling observations is gymnoside I’s procoagulant effect in heparin-induced anticoagulated mice. Heparin sodium prolonged TT and reduced TXB2, PL, and FIB levels, while gymnoside I treatment reversed these changes, shortening TT and elevating TXB2, PL, and FIB. These results suggest dual mechanisms: promoting fibrin formation and stimulating platelet activation. The increase in TXB2 further supports platelet signaling involvement. TXB2, PL, FIB, and TT are critical markers for coagulation assessment.21-25 The dose-dependent response indicates pharmacological relevance, with high-dose gymnoside I showing efficacy comparable to protamine sulfate. These findings highlight gymnoside I’s potential as a natural procoagulant agent and underscore the importance of integrating multiple biomarkers to evaluate efficacy. Nevertheless, limitations should be noted: current results are based on animal models, further disease-specific validation is required, and long-term safety data remain unavailable.
The incorporation of PK–PD modeling adds a critical mechanistic layer, enabling quantitative understanding of gymnoside I’s systemic exposure and pharmacological effects. In TCM, where compounds often exert multi-target and time-dependent actions, PK–PD modeling bridges pharmacokinetics with therapeutic outcomes. 26 Observed hysteresis loops, counterclockwise for TXB2 and clockwise for PL, suggest indirect mechanisms, delayed receptor engagement, or metabolite-mediated pathways, consistent with recent pharmacokinetic studies. 27 By quantifying parameters such as IC50, E0, and Ke0, PK–PD analysis informs dosing strategies and helps identify pharmacodynamic targets. Different model types, direct link, effect compartment, indirect response, and mechanism-based, offer distinct advantages.28-30 For example, the effect compartment model explains delayed TXB2 effects, while the indirect response model may capture PL regulation. The mechanism-based model, though complex, provides systems-level insights into multi-target agents typical of TCM. In summary, PK–PD modeling enhances interpretability of gymnoside I’s pharmacological profile and supports rational dose optimization. Nevertheless, limitations remain: current findings rely on animal models, data are preliminary, long-term safety requires further study, and cell-level PK–PD modeling has not yet been conducted to determine gymnoside I’s behavior within target organ cells. Future research should address these gaps.
From the perspective of previous research, most conventional hemostatic agents have been characterized by narrow mechanisms of action, typically targeting a single coagulation factor or platelet activation pathway. In contrast, gymnoside I exhibits a multi-target pharmacological profile, modulating key nodes within both coagulation and inflammatory networks. The identification of TNF, AKT1, and SRC suggests a coordinated therapeutic mechanism that transcends traditional single-target paradigms. These targets are integral components of signaling pathways such as PI3K-Akt, MAPK, and ErbB, which have been implicated in diverse pathological processes. Notably, Liu Chunhua et al.’s study on B. striata highlighted these pathways as common therapeutic axes across multiple disease contexts, reinforcing their relevance in systems-level drug action. 31
The application of network pharmacology in this study provides a comprehensive framework for elucidating the therapeutic potential of gymnoside I. Mapping predicted targets onto hemorrhage-related gene networks identified 26 intersecting genes across purpura, cerebral hemorrhage, and aplastic anemia, supporting a systems biology interpretation of drug action. Network pharmacology is particularly well-suited to the study of natural products and traditional medicine-derived small molecules, which often engage multiple targets and exhibit pleiotropic effects.32,33 It enables the integration of pharmacogenomic data, disease ontologies, and protein–protein interaction networks to construct mechanistic hypotheses that reflect the holistic therapeutic strategies inherent to traditional medicine. Beyond target identification, network pharmacology plays a pivotal role in exploring the concept of shared therapeutic mechanisms across distinct disease states. Many natural compounds exert their effects by modulating fundamental biological processes—such as inflammation, oxidative stress, or immune regulation—that are common to multiple pathologies. This systems-level approach allows researchers to identify overlapping molecular targets and signaling pathways, thereby supporting the development of interventions with broad-spectrum efficacy. In the case of gymnoside I, the identification of key targets such as TNF, AKT1, and SRC across three hemorrhagic conditions suggests that the compound may act on central regulatory nodes relevant to diverse clinical manifestations. More broadly, network pharmacology supports multi-disease modulation, systematic evaluation of multi-component formulations, and modernization of traditional practices. Nevertheless, limitations remain: reliance on degree centrality may overlook other important genes, and cell-level validation of gymnoside I’s effects within target organ cells has not yet been conducted. Future studies should address these gaps to strengthen mechanistic clarity and translational relevance.
To substantiate the computational predictions, dual validation of target engagement was performed using molecular docking and BLI. Molecular docking provides structural insights into the binding affinity and orientation of gymnoside I within the active sites of target proteins, offering a theoretical basis for interaction specificity. 34 It enables the prediction of key binding residues, hydrogen bonding patterns, and hydrophobic interactions, which are critical for understanding the molecular determinants of pharmacological activity. However, docking alone cannot capture the dynamic nature of molecular interactions or account for conformational flexibility under physiological conditions. BLI complements this by measuring real-time binding kinetics, including association and dissociation rates across varying concentrations, under near-native conditions.35,36 The use of BLI in this study confirms the direct interaction of gymnoside I with TNF, AKT1, and SRC, thereby enhancing the credibility of the proposed mechanisms and providing quantitative evidence of binding strength and stability. In the context of natural product and traditional medicine research, the combined application of molecular docking and BLI represents a powerful strategy for validating drug–target interactions. While docking offers rapid, high-throughput screening of potential targets and structural hypotheses, BLI provides empirical confirmation and kinetic characterization, bridging the gap between in silico predictions and experimental reality. This dual approach is particularly valuable for TCM-derived small molecules, which often exhibit complex pharmacodynamics and engage multiple targets with varying affinities. By integrating structural modeling with biophysical validation, researchers can more accurately delineate the molecular basis of therapeutic effects, identify lead compounds with optimal binding profiles, and refine structure–activity relationships. Moreover, the synergy between docking and BLI enhances the translational relevance of TCM research. 37 It supports the modernization of traditional remedies by providing mechanistic clarity and reproducible evidence, which are essential for regulatory approval and clinical development. For gymnoside I, this comprehensive strategy underscores its potential as a systems-level modulator of hemostasis and inflammation, offering new avenues for the treatment of complex hemorrhagic disorders. More broadly, the integration of computational and experimental techniques exemplifies a rational and scientifically rigorous approach to natural product discovery, paving the way for the development of multi-target therapeutics grounded in traditional knowledge yet validated by modern molecular science. 38
Taken together, this study not only advances the pharmacological understanding of gymnoside I but also exemplifies a modern, integrative approach to natural product research. Future investigations should explore structural analogs to optimize efficacy, conduct disease-specific in vivo trials, and assess long-term safety profiles. Additionally, omics-based analyses could further elucidate downstream signaling effects, while formulation studies may improve bioavailability and clinical applicability. By bridging traditional medicinal concepts with contemporary pharmacological methodologies, this work lays a strong foundation for the clinical development of gymnoside I in hemorrhagic disease therapy.
5. Conclusion
This study provides a comprehensive evaluation of gymnoside I, demonstrating its therapeutic potential in a heparin-induced coagulation disorder model. Gymnoside I significantly improved key coagulation indicators, including PL, TXB2, TT, and FIB levels. Pharmacokinetic–pharmacodynamic modeling revealed time-dependent effects and hysteresis loops, suggesting indirect mechanisms of action and possible involvement of active metabolites. Network pharmacology analysis identified TNF, AKT1, and SRC as central targets, implicating multiple signaling pathways related to coagulation and inflammation. The overlap of these targets across different hemorrhagic conditions supports the compound’s potential for multi-disease modulation. Molecular docking and biolayer interferometry further confirmed direct interactions with key proteins, enhancing the credibility of the proposed mechanisms. Together, these findings highlight gymnoside I as a promising multi-target natural compound and demonstrate the value of integrating experimental pharmacology, computational modeling, and molecular validation in the study of traditional medicine-derived therapeutics.
Footnotes
Acknowledgments
The authors acknowledge Prof. Zuying Zhou and Prof. Lin Zheng, colleagues at the State Key Laboratory of Discovery and Utilization of Functional Components in Traditional Chinese Medicine, Guizhou Medical University, for their valuable technical support, which greatly contributed to the successful completion of this study.
Ethical Considerations
This study was approved by the Animal Care and Welfare Committee of Guizhou Medical University (Ethics Approval No. 2403708).
Consent to Participate
There are no human subjects in this article and informed consent is not applicable.
Author Contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Guizhou Science and Technology Department, grant number ZK[2023]126.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data for this study are in the body of the document.
Statement of Human and Animal Rights
All of the experimental procedures involving animals were conducted in accordance with the institutional Animal Care guidelines of Guizhou Medical University, China.