
Abstract
Introduction
Many vitamin D analogs and sterols-related compounds have been identified and extracted from several plants including Solanaceae species. The aim of our study was to evaluate the antifungal potential of 15 vitamin D, D3 analogs, and some sterols-related compounds that have been detected in some plants along with 3 standard antifungal drugs, Isavuconazole, Fluconazole, and Voriconazole, on the Mucormycosis-sterol 14-alpha demethylase (CYP51) enzyme by in silico study.
Methods
In this work, the molecular simulation was used to prepare the specific enzyme and the antifungal profiles utilized for the molecular docking of the contemplated compounds and subsequent prediction for the mechanism of binding within the catalytic site of Sterol 14-Demethylase (CYP51) to speculate their binding affinities. Anti-fungal medications, Fluconazole and Voriconazole revealed drugs bound distant from the binding sites, but Isavuconazole bound close to the binding site. In this context, the 15 vitamin D and D3 compounds were somewhat seen in binding proximity, especially for Alfacalcidol, Eldecalcitol, and Inecalcitol.
Results
Seocalcitol and Isavuconazole had the lowest binding energies and were the closest ones connected to the binding cavities with the best binding free energies equal −13.21 ± 0.24 and −15.29 ± 0.25, respectively.
Conclusion
Isavuconazole and Seocalcitol could potentially be further assessed to be adjuvantly used as drugs to inhibit the fungal activity by targeting CYP51, a significant target for anti-fungal as well as anti-protozoan drugs, and hence could be efficient against mucormycosis.
Introduction
Mucormycosis, the so-called black fungus, is a parlous infection and needs to be treated with antifungal medicine, usually amphotericin B, posaconazole, or isavuconazole. These medicaments are either given intravenously like amphotericin B, posaconazole, isavuconazole, or by oral administration like posaconazole, isavuconazole. Other drugs, including voriconazole, fluconazole, and echinocandins, do not target Mucoraceae. Often, mucormycosis needs surgical intervention for tissue debridement at the site of infection. Following candidiasis as well as aspergillosis, “mucormycosis” is considered to be the least prevalent species of fungal infection. Mucormycetes, which are belonging to the order Mucorales, cause a rare but serious opportunistic infection. Mucormycetes mold may be found in many places including leaves, rotting wood, manure, soil, and much more other spots. The disease is caused by Mucoraceae species such as Mucor racemosus, Rhizopus pusillus, Absidia elegans, Rhizopus arrhizus, and Apophysomyces elegans.1–3
Mucormycosis can potentially cause vascular thrombosis and tissue necrosis. According to previous studies, the most prevalent variant is rhino-cerebral mucormycosis. Uncontrolled diabetes mellitus as well as leukemia are the most important risk factors for its development. It would be fatal if spread to the CNS during the progress of rhino-cerebral Mucormycosis. The mortality rate could reach up to 60% in case of lung infection which represents the second most common site of infection along with nasal sinuses.4–6
There is an inclusive grasp that vitamin D2 is considered a plant form of vitamin D. 7 Previous studies on many plants including C. diurnum, S. glaucophyllum, T. flavescens and S. glaucophyllum, as well as Arabidopsis thaliana, had led to Vitamin D3 and related compounds detection. In addition, Vitamin D3 relevant compounds and its precursor 7-dehydrocholesterol have been extracted and detected in the leaves of many plants, particularly those belonging to Solanaceae species. 8 The ratio of campesterol to sitosterol that modulates growth in Arabidopsis is controlled by Sterol Methyltransferase 2. 9 Sterols are precursors of numerous steroid hormones encompassing vitamin D. Steryl esters are found in all plants, and overwhelmingly present in the plant cells’ cytoplasm. 10 The hydroxy group of sterols at position 3 can also be bound to a sugar-producing what so called a steryl glycoside. Solanaceae family plants reveal an unparalleled plenty of these glycosides. 11 Campesterol, sitosterol, and stigmasterol are the most abundantly predominators among more or less 250 sterols that had been detected in plants.12,13
In a severe COVID-19 condition, the immune system of infected patients may become dysfunctionally affected, resulting in lymphocytopenia and a dramatic increase in immune-mediated cytokines which can lead to pulmonary hyperinflammation as well as death in some patients. In severely ill patients, clinicians preferred to employ immunosuppressants or steroids as a life-saving therapy depending on the severity of hyperinflammation rather than viral load. A steroid decreases inflammation in the lungs, but it also lowers the body's immunity and raises blood sugar levels in diabetic and non-diabetic people.14–16 In this case, patients who have been immunosuppressed, due to steroids, are more likely to get Mucormycosis or Black fungus.17,18
In addition, diabetes mellitus exacerbates the likelihood of the COVID-19 course severity and is linked to an increase in in-hospital mortality. Diabetes, which is linked to Mucormycosis infection and a significantly raised risk of SARS-CoV-2 infection, could have ominous effects on the surroundings.19–22
Amphotericin B was determined to be the chosen antifungal agent of therapy for the majority of Mucormycosis infections in various investigations. Nevertheless, the duration of treatment is not accurately defined by physicians. According to some studies, at least three weeks of Amphotericin B treatment is recommended, and if radiological as well as clinical improvement is seen, additional treatment with triazoles including Posaconazole, Isavuconazole, Voriconazole, and others is recommended.23–26 According to researchers, Posaconazole is the most used alternative to Amphotericin B for the treatment of Mucormycosis infection. Animal studies show that posaconazole is more efficacious than itraconazole but less effective than amphotericin B. Posaconazole medication bioavailability is increased whether administered intravenously or by tablet. Itraconazole, a broad-spectrum triazole with significant in-vitro efficacy against Mucorales, has been shown to demotivate Mucorales in clinical studies. In an in-vitro model, voriconazole was found to be ineffective against Mucorales. As a result, triazoles should not be used as a first-line treatment for Mucormycosis.27,28 Marty et al reported that Isavuconazole is well-tolerated and is as efficient as amphotericin B and could be utilized for the treatment of mucormycosis. 29 Poply et al. reported that low serum Vitamin D concentrations are supposedly embroiled as a typical risk factor for the incidence of mucormycosis in patients with COVID-19 and giving calcitriol supplements to retain normal serum concentrations could possess a positive impact on this issue. 30
There are sundry conceivable mechanisms by which vitamin D can combat and resist infection like inducement of the expression of defensin and cathelicidin. Moreover, vitamin D and related sterols can shift and switch the pro-inflammatory response spent by Th1 and Th17 to the anti-inflammatory bounce exerted by Th2 and Treg so as to suppress cytokine storms. 31
Lately, computational research tools have become a pivotal component in drug discovery practicability. Molecular modelling is typically utilized delineating method for striking recognition gadgets when the 3D − crystal structure of both ligands and studied molecules are renowned.32–41
This study aimed to evaluate the potential antifungal efficacy of 15 vitamin D and D3 analogs along with 3 standard antifungal drugs, Isavuconazole, Fluconazole, and Voriconazole, on the Mucormycosis-sterol 14-alpha demethylase (CYP51) enzyme by molecular docking tools.
Materials and Methods
The primary method used to proceed with this research study is a literature review. After reviewing the literature, the dataset was used for conducting this research study.
Dataset
The three-dimensional (3D) structural entity of sterol 14-alpha demethylase from C. albicans and some other fungi commonly abbreviated as CYP51 was regained from the database of Protein Data Bank (PDB). 42 The CYP51: PDB ID: 5TZ1 is experimentally modelled by the X-RAY Diffraction method with a resolution of about 2.00 Å. The compounds as ligands were retrieved into two sets. One set consists of standard compounds as these compounds are FDA-approved drugs (three drugs) and the other set consists of 15 compounds. Those compounds are synthetics, derivatives, and analogs of Vitamin D and Vitamin D3. 43
Structure-Based Virtual Screening
The structure-based virtual screening technique which is used to design drugs computationally in the early stages of drug discovery. In this technique, the compounds in the presented study (standard 3 drugs as controls and 15 compounds of Vitamin D, D3 analogs and derivatives) were selected against a target for the drug. The 3D structure of CYP51: 5TZ1 was obtained to bind a total of 18 compounds into the binding pocket. The compounds were selected based on identified binding scores as binding energies and suitable poses for further analysis. 44
Molecular Docking
The computational approach of molecular docking (Protein-ligand) was employed to achieve the best suitable conformation, identify the binding region of CYP51, and predict a reliable interaction between CYP51 protein and ligands (3 controls and 15 Compounds). The docking was done by using computational tools such as AutoDock Vina. 38 AutoDock is a set of automated docking algorithms. AutoDock VINA algorithm is designed to predict the binding of small molecules as drug candidates with the receptor (target protein). It is a suite of automatic docking programs that are based on the Lamarckian genetic algorithm (LGA) technique. This tool was used to identify the suitable binding location and conformation of the compound. AutoDock Vina expressively improves the accuracy of interacting mode calculations.
Preparations for Molecular Docking
Energy Minimization
The receptor CYP51 and ligands (3 drug controls and 15 Compounds) were subjected to an energy minimization process for the removal of their energy constraints. The UCSF Chimera 44 tool was utilized for energy minimization of receptors and ligands. The ligand and receptor structures were minimized by utilizing the algorithm's steepest descent: 100 and conjugate gradient: 100 steps. By assigning the charges to minimization for standard residues AMBERff14SB and other residues AM1-BCC. The charges were computed by using ANTECHAMBER and AM1-BCC methods.
Receptor and Ligand Preparation
In AutoDock Vina analysis, receptor molecules were prepared by adding polar hydrogen atoms and Kollman charges. For ligands, Gasteiger partial charges were allocated, and non-polar hydrogen atoms were merged. The docking experiments were performed with a receptor in a rigid state and ligands as flexible by permitting all torsions to rotate. Docking runs were carried out with a grid map and spacing in Angstroms (Å). The grid dimensions as grid center (X: 62.5942, Y: 66.7693, Z: 2.6613) and dimensions (X: 66.5549579811, Y: 55.0975105667, Z: 67.9245097923). The empirical free energy function and Lamarckian genetic algorithm (LGA) were applied. The remaining docking parameters were set to default.
Docking Run
After preparations, the docking was carried out and predictively, the results were clustered according to RMSD criteria. The perfect docked conformations of ligands were selected based on binding free energy values to evaluate the binding of CYP51 protein with 3 drug controls and 15 compounds. The suitable docked complexes for each ligand from two sets were selected and interactions were analyzed by using Discovery Studio 45 which generated a plot of interactions too. The Interactions were also analyzed by using UCSF chimera 46 which is a visualization tool.
Molecular Dynamics (MD) Simulation Assays
The stability of top-selected compounds and their binding energies as well as the molecular dynamics simulation were carried out. The Gromacs software 47 was utilized for simulations.
GROMACS
The Gromacs software contains a pre-processor, molecular dynamics and energy minimization program which can be used by random processors and many other analysis algorithms. GROMACS stands for Groningen Machine for Chemical Simulation, developed in the early 1990s at the University of Groningen. Gromacs is flexible, fast, and free-of-cost server for MD simulations, which is well-suited against many force fields such as OPLS, GROMOS, ENCAD, and AMBER. 48
Preparation of Input Files
Possibly the residues or atoms may be missed during minimization or dockings. Therefore, receptor or ligand pdbs were checked for missing residues and repaired by AutoDock and saved the files to run MD simulations. The best binding poses of the Isavuconazole and Seocalcitol with CYP51 protein were selected from the molecular docking studies and subjected to 100 ns MD simulation in order to analyze the protein-ligand stability. The protein-ligand complexes of both systems were solvated in a cubic box of 10 Å containing TIP3P water molecules. 49 K + and Cl- counter ions were added to neutralize the systems. To exclude the steric clashes, after neutralization, the systems were lessened by the steepest decent technique for 5000. Thereafter, both systems were equilibrated at NVT and NPT ensemble for 50000, and 100000 steps, respectively, at 310 K temperature to adjust the systems for the production run. 50
Simulation Runs
The simulations in this study utilized the Berendsen thermostat and Parrinello-Rahman algorithms to maintain a constant temperature of 310 K and pressure of 1 atm. The relaxation of the systems was conducted with a time step of τ T = 0.1 ps for temperature relaxation and τ P = 2.0 ps for pressure relaxation. The LINCS algorithm was employed to maintain optimal bond lengths for hydrogen atoms, while the Verlet algorithm was used for calculating nonbonded interactions.51,52 The Particle Mesh Ewald method was implemented to account for electrostatic interactions beyond the short-range cutoff. 53 Periodic boundary conditions were applied in all three dimensions (x, y, and z), and the systems underwent a production run. Trajectories from the production run were stored at intervals of 10 ps and analyzed using Gromacs commands and the BIO3D package in R. 54 The simulations were performed using the Gromacs simulation package and the CHARMM36 forcefield.55,56
Post-Simulation Analysis
After the completion of the molecular dynamics (MD) simulations, the behaviour of protein was analyzed with the passage of time as well as the binding stability of complexes. Analyses of numerous dimensions were performed as RMSD, RMSF, PCA, and MMGBSA.
Results
CYP51 belongs to CYP (Cytochrome P45051) family. It involves in ergosterol synthesis (fungal-specific). C. albicans causes widespread fungal infections in humans, while CYP51 is the target for antifungal drugs to treat fungal infections.40,57,58
Secondary and Three-Dimensional Structure Analysis of CYP51
To achieve the goal of this research study, the 3D structure of CYP51: 5TZ1 was retrieved from PDB database.42,45 The protein sequence was examined from UniProt database (UniProt consortium, 2015) and protein uniport ID: P10613. CP51_CANAL.The sequence length of CYP51 is about 528 AA and 60,675 mass in Dalton. To infer the secondary structural information the PDBsum59,60 was utilized while for 3D structure insights the Chimera visualization tool was employed (Figure 1B). By analyzing the 2D structure of CYP51 (5TZ1) it was observed that there are 5 Beta sheets, 4 beta hairpins, 1 beta bulge, 14 strands, 22 helices, 42 helix-helix interacts, 35 beta turns, and three gamma- turns (Figure 1A).

Representation of 2D and 3D structure of CYP51: (A) The 2D structure of CYP51 is represented in purple color (Helices), Orchid color (Strands with their sheets), and dark purple (loops). The sequence of CYP51 is shown in single residue alphabet (blue color) with length numbering in black color. Beta hairpins with sheets labelling are illustrated in red color while the gamma turns and beta turns are labelled in orchid color. (B) The 3D structure of CYP51 is represented in purple (Helices), orchid (Beta sheets), (Coils) dark purple colors. The labelling of Helices is in blue.
Drugs (Controls) and Compounds as Ligands
In this research study, the three compounds are considered as controls while the other 15 compounds are the synthetics, derivatives, and analogs of Vitamin D and Vitamin D3. The three drugs namely Isavuconazole, Fluconazole, and Voriconazole. The other 15 compounds are listed as Alfacalcidol, Calcifediol, Calcipotriol, Calcitriol, Cholecalciferol, Doxercalciferol, Eldecalcitol, Ergocalciferol, Falecalcitriol, Inecalcitol, Paricalcitol, Seocalcitol, Tacalcitol, Vitamin D3, and Oxacalcitriol. These compounds were obtained from PubChem while some of them are natural compounds and some are synthetics (Table 1). The structures of these drugs and compounds are shown in Figures 2 and 3.

2D structures of 15 compounds fetched from PubChem. (A). Alfacalcidol, (B). Calcifediol, (C). Calcipotriol, (D). Calcitriol, (E). Cholecalciferol, (F). Doxercalciferol, (G). Eldecalcitol, (H). Ergocalciferol, (I). Falecalcitriol, (J). Inecalcitol, (K). Paricalcitol, (L). Seocalcitol, (M). Tacalcitol, (N). Vitamin D3, and (O). Oxacalcitriol.

2D structures of Drugs. (A). Isavuconazole, (B). Fluconazole and (C). Voriconazole.
The Fetched 15 Compounds From PubChem Database With Their Description.

Isavuconazole
The prodrug, water-soluble triazole named Isavuconazole has an antifungal activity widely. It inhibits the CYP51 which catalyzes the lanosterol into ergosterol conversion. Isavuconazole absorbs easily and is given both intravenously and orally. They were initially approved in 2015 by the U.S. FDA (Food as well as Drug Administration). 61
Fluconazole
Synthetic triazole which contains antifungal action termed Fluconazole. Fluconazole is the approved medicine by U.S. FDA as an antifungal prescription. It is usually prescribed to treat esophageal candidiasis, vulvovaginal candidiasis, and oropharyngeal candidiasis. 61
Voriconazole
The synthetic triazole compound has an antifungal mode of action termed Voriconazole. It inhibits the demethylation of 14-alpha-lanosterol. Voriconazole is also an approved prescription against fungal infections such as invasive candidiasis and esophageal candidiasis by the U.S. FDA. 61
Molecular Docking Analysis
To reveal the fungal activity of CYP51 (5TZ1), the binding of controls and 15 compounds against the target CYP51 was done by using the Molecular docking technique. This study is a virtual study of binding assays without any physico-chemical measurements. The docked complexes were analyzed by utilizing Discovery Studio and Chimera. The three controls were observed in complex form as bound in the binding regions of the protein CYP51. The binding affinities were also calculated for these complexes. The binding affinity for Fluconazole: −7.4, Voriconazole: −7.6, and Isavuconazole: −8.5 (Figure 4). The lowest binding affinity shows the suitable accurate binding between the ligand and receptor. The lowest binding affinity was observed for Isavuconazole. The drug Isavuconazole and Voriconazole were docked in the binding region similarly with minor differences while the Fluconazole was observed as far away from the binding region (Figure 5).
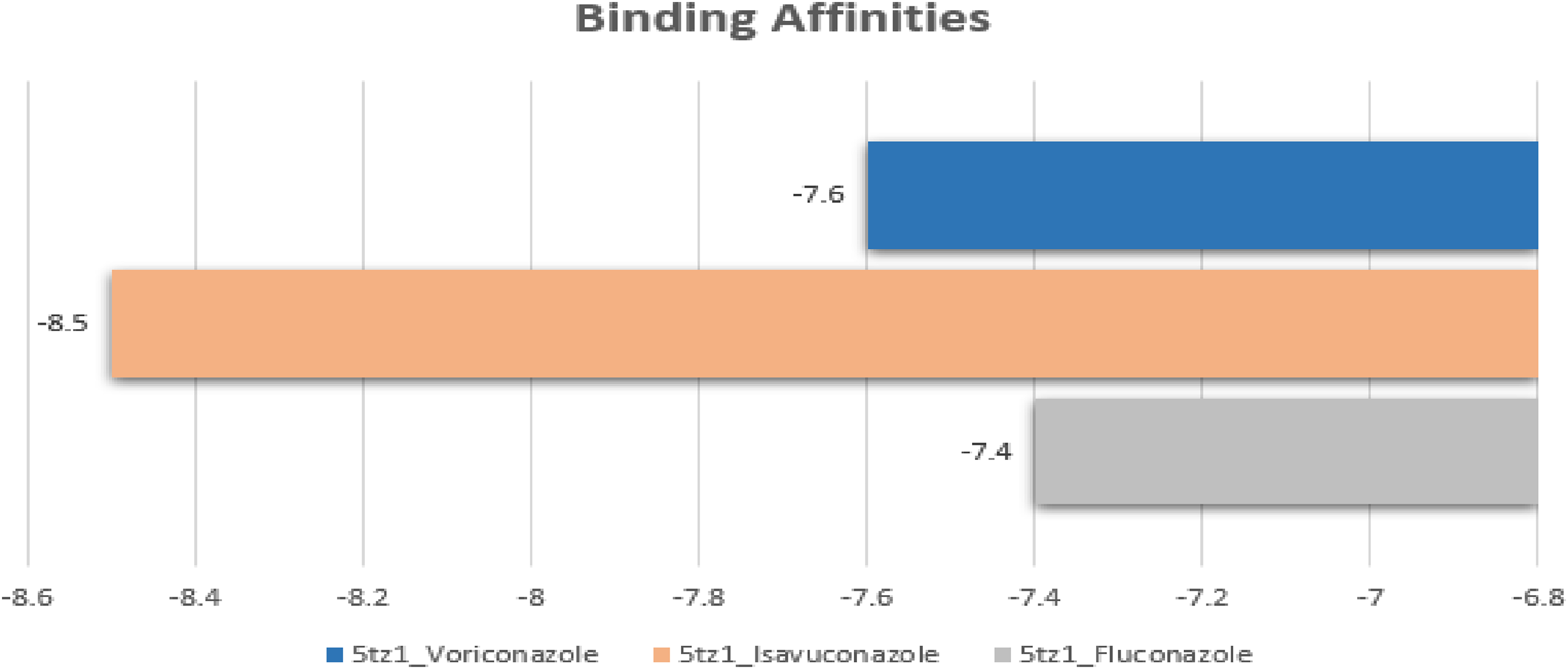
Binding affinities of three drugs (Controls). The binding affinities of controls docked complexes are shown in a bar graph. The CYP51-Fluconazole is shown in gray color with weak binding affinity. The CYP51-Voriconzole and CYP51- Isavuconazole are represented in blue and orange colors while the orange bar is showing highest value while blue bar as higher than gray but lower than orange. The most suitable binding affinity is of CYP51-Isavuconazole.

Interaction analysis of three drugs (Controls). (A). The 2D diagram of CYP51-Isavuconazole interaction. The Isavuconazole is shown in chartreuse color with ball and stick representation. While interacting residues are shown in different colors according to their force of interactions. (B) The 2D diagram of CYP51-Voriconazole interaction. The Voriconazole is represented in ball and stick depiction (Orange red) while the interacting residues are shown by ball representation in different colors. (C) The protein CYP51 is depicted in white color with mesh surface representation while the three drugs (Chartreuse, Orange red, and blue) are bound in different regions. (D) The 2D diagram of CYP51-Fluconazole interaction. The Fluconazole is depicted in blue color with ball and stick representation. The residues involved in the interaction are represented in ball with different colors. The different colors for all interactions are presented as Light green: Vander Waals, Green: Conventional hydrogen bonds, Pink/Plum: Alkyl/Pi-Alkyl, Light sky blue: Carbon-hydrogen bond, Red: Unfavourable donor-donor, Sky blue: Halogen (Fluorine), Hot pink: Pi-Pi stacked and Magenta: Amide-Pi stacked.
The interacting residues were observed in binding between CYP51 and controls (Figure 5). As CYP51-Fluconazole: Tyr64, Gly65, Leu87, Leu 88, Lys90, Leu121, Pro230, Phe233, Val234, Phe300, Leu376, His377, Ser378, Tyr505, Ser506, Ser505, Met508, Val509, CYP51-Voriconazole: Thr122, Ile131,Phe126, Leu139, Tyr132, Lys143, Gln142, Leu300, Phe228, Ile304, Gly303, Gly308, Gly307, Thr311, Ile471, Gly472, Leu376 and CYP51-Isavuconzole: Tyr118, Phe126, Ile131, TYyr132, Leu139, Lys143, Leu150, Leu204, Ile304, Gly307, Gly308, Leu376, Ile379, Arg381, His468, Cys470, Ile 471, Gly472, Phe475. The differences were seen in the binding region where three drugs were bound. The Fluconazole was observed as completely out of the binding vicinity while Voriconazole and Isavuconazole were observed in the same region to some extent but showed differences in interacting residues. While considering the binding affinities of all three drugs, the most suitable drug as Isavuconazole was selected for further analysis and comparison against other compounds because of its lowest binding affinity and most relatable interacting residues (Binding vicinity).
The natural, synthetic, analogs and derivatives of Vitamin D and Vitamin D3 compounds were docked against CYP51 to inhibit the fungal activity with regard to controlling the fungal infections. The docked complexes were analyzed and visualized by Chimera and Discovery Studio. The binding affinities were observed as CYP51-Alfacalcidol −9.8, CYP51-Calcifediol −9.9, CYP51-Calcipotriol −9.5, CYP51-Calcitriol −10.0, CYP51-Cholecalciferol −9.7, CYP51-Doxercalciferol −10.1, CYP51-Eldecalcitol −7.8, CYP51- Ergocalciferol −9.8, Falecalcitriol −10.1, CYP51-Inecalcitol −9.8, CYP51-Oxacalcitriol −9.0, CYP51- Paricalcitol 10.2, CYP51- Seocalcitol −10.4, CYP51-Tacalcitol −9.5 and CYP51-Vitamin D3 −9.1. The binding affinities are also shown in graphical representation as in bar graph (Figure 6).

Binding affinities of all 15 compounds: The binding affinity for each complex is shown in different 15 colors. The labelling of each bar is represented with a similar color to the bar color. The complex with the lowest binding affinity is depicted in cornflower blue color (5TZ1-Seocalcitol: 10.4). The binding affinities are also labelled in white color.
All binding affinities were suitable as they fall in between −7.8 and −10.4. The minimum binding affinity also lay in the good range while after analyzing all the binding affinities, it was depicted that all these 15 compounds strongly bound with the target protein CYP51 but varied in the binding cavity. Based on binding affinities (lowest binding affinity), the one complex was selected as a top complex from 15 compounds which is CYP51-Seocalcitol with binding energy −10.4.
The 15 compounds (Vitamin D and D3 analogs, derivatives) were docked against Sterol 14-Demethylase (CYP51) to identify the most potent new inhibitors other than many traditional compounds such as imidazoles, triazoles, and tetrazoles while to confirm the potential binding sites of CYP51, the previous research study took as reference. The 15 complexes were examined regarding their binding affinities as well as their binding sites (Figure 7).
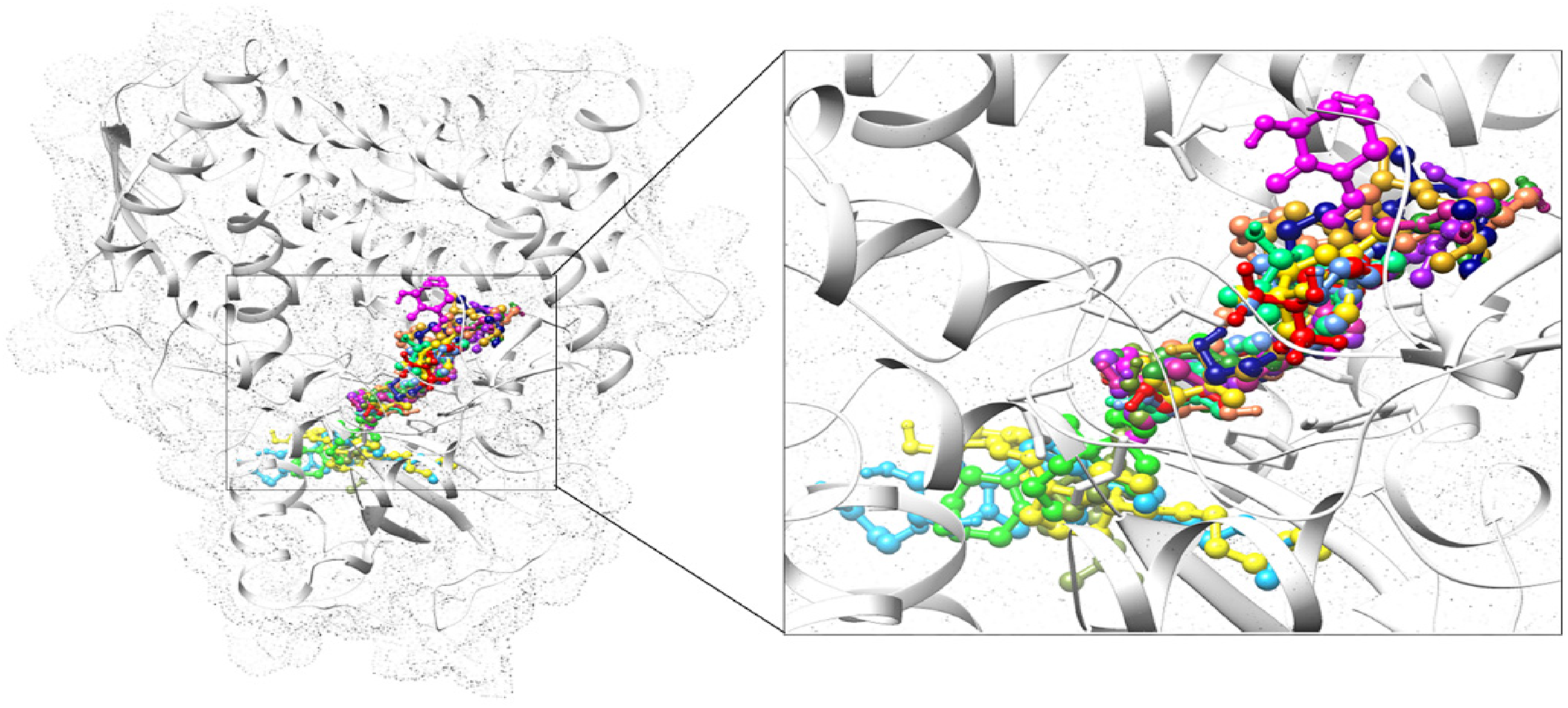
Interaction visualization of 15 compounds with CYP51. The target protein CYP51 is illustrated in white color with a dot surface representation. The 15 compounds are shown in bound form in their specific binding region. The 15 compounds are depicted in different colors as Alfacalcidol: Yellow, Calcifediol: Purple, Calcipotriol: Spring green, Calcitriol: Violet red, Cholecalciferol: Navy blue, Doxercalciferol: Coral, Eldecalcitol: Deep sky blue, Ergocalciferol: Goldenrod, Falecalcitriol: Red, Inecalcitol: Olive drab, Paricalcitol: Fresh green, Seocalcitol: Magenta, Tacalcitol: Cornflower blue, Vitamin D3: Green, Oxacalcitriol: Gold.
The 11 compounds were bound in the binding vicinity while the other four compounds were completely observed beyond the binding vicinity. The Calcipotriol, Calcitriol, Cholecalciferol, Doxercalciferol, Ergocalciferol, Falecalcitriol, Paricalcitol, Seocalcitol, Tacalcitol, and Oxacalcitriol were observed in the binding vicinity. The interacting residues for some of these complexes were observed as similar to reported binding sites while some complexes showed few similar interacting residues as with reported ones. The Alfacalcidol, Eldecalcitol, Inecalcitol, and Vitamin D3 complexes interacting residues are more different from the reported ones, and the overall binding region for these was also observed beyond the binding sites. So, the top complex was selected based on the lowest binding affinity and similarity in the binding region (interacting residues) with already reported data. The top complex from 15 compounds was selected as CYP51-Seocalcitol while from 4 controls the CYP51-Isavuconazole.
The binding affinities of 15 compounds in complexes were identified and shown in Figure 6 while the interaction analysis was also done and shown by interacting 2D diagrams. All 2D interaction diagrams are shown in Figure 8.

Interaction analysis of 15 compounds with CYP51. The 2D diagram interaction analysis of 15 compounds against CYP51. The compounds are represented in different colors with ball and stick representation, Alfacalcidol: Yellow, Calcifediol: Purple, Calcipotriol: Spring green, Calcitriol: Violet red, Cholecalciferol: Navy blue, Doxercalciferol: Coral, Eldecalcitol: Deep sky blue, Ergocalciferol: Goldenrod, Falecalcitriol: Red, Inecalcitol: Olive drab, Paricalcitol: Fresh green, Seocalcitol: Magenta, Tacalcitol: Cornflower blue, Vitamin D3: Green, Oxacalcitriol: Gold. The target protein CYP51 interacting residues are illustrated in their specific colors based on forces of interactions. The diverse colors for all interactions are depicted as Light green: Vander Waals, Green: Conventional hydrogen bonds, Pink/Plum: Alkyl/Pi-Alkyl, Light purple: Pi-Sigma, Light sky blue: Carbon-hydrogen bond, Red: Unfavourable donor-donor, Unfavourable acceptor-acceptor, Sky blue: Halogen (Fluorine), Hot pink: Pi-Pi stacked and Magenta: Amide-Pi stacked.
Comparative Analysis Between Potent Top Complexes
The CYP51-Isavuconazole and CYP51-Seocalcitol were nominated as top complexes from drugs and 15 compounds, respectively (Figure 9). These two complexes were selected on the basis of the lowest binding affinities as well as similarities in interacting residues between reported data (Binding regions) and our generated results.

Top complexes comparison. (A) The interaction analysis of Seocalcitol with CYP51. The compound is shown in magenta color (Ball and stick representation) while the interacting residues of CYP51 are represented by their specific interaction colors. (B) The interaction analysis of Isavuconazole with CYP51. The compound is represented in chartreuse color with ball and stick representation while the interacting residues of CYP51 are characterized by their specific interaction colors. (C) The two ligands as Seocalcitol and the drug (Isavuconazole) are shown in a complex form with CYP51 while illustrating their specific binding region in CYP51. The target protein is shown in White color (Mesh surface representation). Both ligands (Seocalcitol and Isavuconazole) are depicted in sphere representation (Magenta and Chartreuse).
The binding cavities which were observed in the selected top complexes are shown in Figure 10. The interacting residues of CYP51 binding cavity which were involved in interaction with Seocalcitol (Tyr64, Leu121,Tyr118, Phe126, Thr122, Tyr132, Ile131, Pro230, Phe228, Ile304, Phe233, Gly307, Gly308, Thr311, Leu376, His377, Ser378, Phe380, Cys470, Gly472, Ala476, Ser507, Met508) and Isavuconazole (Tyr118, Ile131, Phe126, Leu139, Tyr132, Leu150, Lys143, Leu204, Ile304, Gly307, Gly308, Thr311, Leu376, Ile379, Arg381, His468, Cys470, Ile471, Gly472, Phe475). The conserved interacting residues and hydrogen bonds in both complexes are also illustrated in Figure 10.

Comparison between binding cavities of top complexes (CYP51-Isavuconazole and CYP51-Seoalcitol). (A) The complex between CYP51 (white color) and Isavuconazole (chartreuse color). The binding cavity is shown in orchid color whereas the bound Isavuconazole is shown in ball and stick representation. The only binding cavity is depicted in solid surface representation while the other protein is in ribbon form. (B) The complex between CYP51 (white color) and Seocalcitol (magenta color). The binding cavity is exposed in aquamarine color where the bound Seocalcitol is revealed in a ball and stick representation. The only binding cavity is represented in solid surface representation while the other protein is in ribbon form. The common interacting residues of binding cavities in both complexes are labelled in black color while the residues involved in hydrogen bonding are labelled in red color.
Molecular Dynamics Simulation Analysis
The top complexes and best binding pose of Isavuconazole and Seocalcitol with CYP51 protein were selected from the molecular docking studies and subjected to 100 ns MD simulation in order to analyze the protein-ligand stability. The 100 ns MD runs were conducted against these two complexes and post-simulation analyses were performed to confirm the stability in comparison.
Root Mean Square Deviation (RMSD)
To assess the stability of the protein-ligand complex, the root mean square deviation (RMSD) of the C-alpha atoms of the protein and the ligand atoms was calculated throughout the simulation duration (see Figure 11). The analysis revealed that the systems reached equilibrium within the first 10 nanoseconds of the simulation. After equilibration, the RMSD values remained relatively stable, indicating a favourable and consistent interaction between the protein and the ligand, the RMSD of (CYP51) 5tz1-Seocalcitol attained a value of ∼ 0.5 nm that gradually decreased to ∼ 0.35 at 30 ns. The RMSD value deviated to ∼ 0.5 nm at 40 ns and remained in this range till 60 ns and then for a short period it decreased to ∼ 0.45 nm and again attained the previous range. The maximum RMSD value was recorded at 70 ns with a value of ∼ 0.6 nm. The RMSD looked stable in the last part of the simulation. The RMSD of 5tz1-Isavuconazole deviated in the range of ∼ 0.15 to 0.3 nm till 65 ns after being equilibrated at 10 ns and then attained stability in the range of ∼ 0.2 to 0.3 nm till the end of the simulation. similarly, the RMSD of ligand atoms was calculated which showed that Isavuconazole was remained stable throughout the simulation with a small deviation at the end of the simulation while Seocalcitol showed deviations throughout the simulation. In order to get a better idea about the protein-ligand stability, different snapshots of the trajectories were retrieved and aligned. The aligned snapshots of 5tz1-Isavuconazole showed the ligand was stably bound with protein throughout the simulation (Figure 12A) while the ligand in 5tz1-Seocalcitol showed different poses (Figure 12B). But there was no major difference in the stability of complexes. Both complexes show stability.

The RMSD plots are represented the C-alpha atoms of protein complexed with Seocalcitol and Isavuconazole and the atoms of both ligands throughout the simulation time.

The alignment of MD snapshots of complexes.
Root Mean Square Fluctuations (RMSF)
The Root Mean Square Fluctuations (RMSF) analysis provides insights into the flexibility of individual residues within the protein. Higher RMSF values indicate more flexible regions, such as loops, while lower values suggest more rigid residues, such as helices and sheets. The RMSF plots of the studied systems are depicted in Figure 13. In both systems, the initial residues exhibited higher RMSF values, primarily due to the presence of the N-terminal region. Residues 100 to 200 did not show significant fluctuations, indicating their relative rigidity. Minor fluctuations were observed in residues 220 to 270. Notably, in the 5tz1-Seocalcitol complex, major fluctuations were observed in residues 400 to 450, while the other complex did not exhibit such fluctuations. These observations suggest that specific regions of the protein exhibit varying degrees of flexibility, with loops and certain segments showing higher levels of fluctuations. The differences between the two complexes highlight the unique dynamics and structural behaviour of each system. The remaining residues did not show major fluctuations which indicate that the systems remained stable while the fluctuations in 400 to 450 AA (CYP51-Seocalcitol) did not matter because these residues were not involved in the interaction with ligands. So, overall, both complexes were observed as stable.

The plot is represented the root means square fluctuations of protein amino acids complexed with Seocalcitol and Isavuconazole.
PCA
The dynamic behaviour of the protein in both complexes was analysed by Principal Component Analysis, which helps to test the collective motions of MD trajectories. The PCA plots of both complexes are shown in Figures 14A & 14B. In (5TZ1) CYP51-Seocalcitol complex, it was observed that PC1 showed the highest variation of 27.75% than others. While the PC2 and PC3 depicted a variability of 15.41% and 7.64%, respectively (Figure 14A). While in 5tz1- Isavuconazole complex, the highest variation was observed as 30.75% in PC1. Other variations in PC2 and PC3 were 14.57%, and 7.14%, respectively (Figure 14B). The PCA analysis, utilizing simple clustering in the PC subspace, uncovered the conformational changes present within all clusters. Notably, the blue regions exhibited the most pronounced movement, indicating significant flexibility and substantial conformational changes. The white regions displayed intermediate movement, signifying a moderate level of flexibility and conformational variation. Conversely, the red regions indicated less movement and lower flexibility, suggesting a relatively more stable conformation. This color-coded representation effectively illustrates the varying degrees of movement and flexibility observed across different regions of the system.

Principal component analysis of the top 2 complexes. (A) The plot is indicated that 5tz1-Seocalcitol shows a total variation of about 50.8%. (B) 5tz1-Isavuconazole is represented with a total variation of 52.73.
Molecular Mechanics Generalized Born Surface Area (MMGBSA)
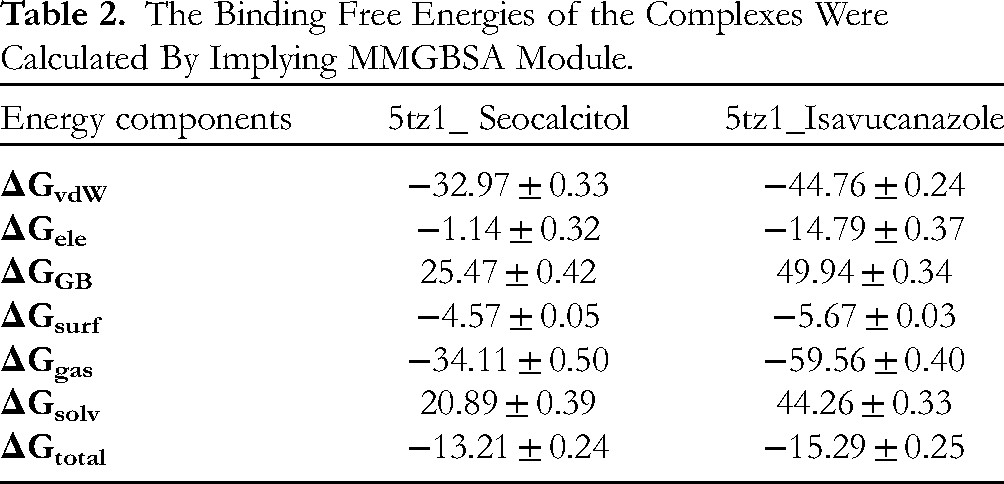
The MMGBSA method was employed to determine the total binding free energy (ΔGtotal) for both complexes. ΔGtotal serves as an indicator of the stability of the protein-ligand complex, with lower values suggesting greater stability and vice versa. It was computed by summing the free energy of the protein-ligand complex and the difference between the CYP51 protein and its ligands’ individual free energies. The MMGBSA model considers various protein-ligand interactions, including van der Waals energy (VDWAALS), electrostatic energy (EEL), and electrostatic contribution to solvation free energy by Generalized Born (EGB). These interactions collectively contribute to the overall binding free energy of the complex. The specific values of the total binding free energies, as estimated using the MMGBSA model, are provided in Table 2. These values offer insights into the strength of the protein-ligand interactions and can be used to assess the relative stability of the complexes under investigation. The result is indicating that both complexes were observed in a stable state. The minor difference in ΔG total between these two complexes was observed at about two points (−13.21 ± 0.24 versus −15.29 ± 0.25). But overall, both complexes were showing stability regarding binding energies (Table 2).
The Binding Free Energies of the Complexes Were Calculated By Implying MMGBSA Module.
Discussion
Drug discovery is confronting a lot of bewilderments to develop a presumed drug for medical intent, hence, to deal with such dilemmas, computational studies are utilized to uphold experimental works. 62
Integrating the chemical, computational, and biological aspects of synthesized molecules is crucial for understanding natural products. By combining these perspectives, researchers can gain insights into structure-activity relationships and potential as well as therapeutic applications. Computational methods help predict molecular properties and behaviour, aiding in screening and prioritizing natural product candidates. Vitamin D-related compounds share a common Secosteroid structure with variations in the steroid backbone and functional groups. These modifications greatly influence their biological activity and properties.63–69
The CYP51 which is also known as ERG11 belongs to the CYP (Cytochrome P450 monooxygenase) superfamily. It facilitates the vital step of ergosterol synthesis (Fungal-specific sterol). The CYP51 is also the target of drugs and inhibitors in clinical trials to stop fungal infections. The inhibition of fungal infections can occur as the weakening of coupled ergosterol by a collection of 14α-methyl sterols which leads to the growth arrest of fungal cells. There are many drugs or inhibitors reported in the recent literature as well as in the market that have anti-fungal efficacy but still need further characterization and recognition. The three-dimensional structure of CYP51 was retrieved as it is considered a target for 2 sets of compounds: Standard compounds (3) and 15 Vit D, D3 analogs (Compounds). The computational technique (Molecular docking) was utilized to identify drug-binding target sites.
In the case of standard compounds, Fluconazole (B.E: −7.4) was detected as out of the binding vicinity while Isavuconazole (B.E: −8.5) and Voriconazole (−7.6) was detected in a similar region with differences in interacting residues. In the case of synthetic, natural, derivatives, and analogs of Vitamin D, Vitamin D3 compounds, the binding energy analysis of all 15 compounds depicted that all these 15 compounds strongly interacted with CYP51 but showed variations regarding binding cavities while the lowest binding affinity is −10.4 of Seocalcitol complexed with CYP51.
In previous studies, the CYP51 protein was reported in complex with other drugs. The different types of compounds or drugs have already been reported in previous research studies as complexed with CYP51 target protein with respect to different organisms. The imidazole compounds such as Miconazole, Clotrimazole, Econazole, and Ketoconazole were tested as drugs. The 2 triazole generations as Posaconazole, Itraconazole, Fluconazole, Voriconazole, and Isavuconazole were also tested in the clinic as drugs. Some new compounds (VT-1611, VT1129, and VT-1598) because of the lowest binding affinity have also been identified. Some other inhibitors (Ravaconazole, Efinaconazole, Sertaconazole, Luliconazole, and Oxiconazole) were also observed as already reported against CYP51 targets to inhibit topical fungal infections. 61
By reviewing the previously reported research studies, it was observably obvious that there are some regions and residues that are involved in interactions with drugs. The six putative regions regarded as Substrate-Recognition-Site 1 (SRS1) to Substrate-Recognition-Site 6 (SRS6) were reported where drugs or inhibitors can bind to inhibit the fungal activity. While SRS1 and SRS4 were reported as the most significant sites in the case of CYP51 target protein. The amino acid positions in sequence are SRS1: 117-134 and SRS4: 305-314. 61
The already (reported) docked complexes were also analyzed regarding their binding residues (CYP51) which were involved in interaction with drugs as CYP51 _ Posaconazole (28 residues) and CYP51_VT1161 (22 residues). 48 The residues are shown in Table 3.
The Involved Residues in the Interaction Between CYP51 And Inhibitors. The Red Font is for Hydrogen Bonds.

Some binding sites were also fetched from UniProt database. The database collected this information through manual assertion which was done by a combination of experimental and computational evidence. The binding sites were noted as Tyr118, Gly307, and His377 in the case of inhibitors Itraconazole, Posaconazole, and Oteseconazole, respectively (UniProt consortium, 2015). The reported binding vicinity in previous research studies as Helix 1 (Phe58, Ala61, Ala62, Tyr64, Gly65), β1-β2 turn (Leu88), Helix 4 (Tyr118, Leu121, Thr122, Phe126), β helical turn (Ile131, Tyr132), Gamma turn, Helix10 (Phe228, Pro230, Phe233), Helix13 (Gly303, Ile304, Gly307, Gly308, Thr311), β1-4 loop (Leu376, His377, Ser378), β1-4 strand (Phe380), and β4 hairpin (Tyr505, Ser506, Ser507, Met508). 61
The binding residues or binding cavities of these reported complexes were considered as a reference to identify the potential binding sites of CYP51 against newly identified compounds. The Calcipotriol, Calcitriol, Cholecalciferol, Doxercalciferol, Ergocalciferol, Falecalcitriol, Paricalcitol, Seocalcitol, Tacalcitol, and Oxacalcitriol interacted in the binding vicinity. The binding residues of some of these complexes were detected as the same as the reported binding sites while few complexes revealed some same binding residues as with reported ones. The Alfacalcidol, Eldecalcitol, Inecalcitol, and Vitamin D3 complexes binding residues are more dissimilar to reported ones, and binding sites for these were also observed beyond the binding vicinity. The CYP51-Seocalcitol and CYP51-Isavuconazole complexes were selected as top complexes for further analysis because of the lowest binding energies as well as similarity in interacting residues with each other and as well as according to the already reported, published research studies. The top complexes binding pockets were revealed as CYP51-Isavuconazole: Tyr118, Phe126, Ile131, Tyr132, Leu139, Lys143, Leu150, Leu204, Ile304, Gly307, Gly308, Thr311, Leu376, Ile379, Arg381, His468, Cys470, Ile471, Gly472, Phe475 and CYP51-Seocalcitol: Tyr64, Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Pro230, Phe233, Ile304, Gly307, Gly308, Thr311, Leu376, His377, Ser378, Phe380, Cys470, Gly472, Ala476, Ser507, Met508. The binding cavities residues were observed as most similar to the binding sites or regions that were fetched from the previously reported data (Table 2). 48 The residues Tyr118, Phe126, Ile131, Tyr132, Ile304, Gly307, Gly308, Thr311, Leu376, Cys470, Gly472 were observed as common in both complexes. The hydrogen bonds were also detected as Thr311 and Tyr132 in CYP51-Seocalcitol and CYP51-Isavuconazole, respectively. While His377 was observed in previously reported data by Zhang et al. 61 Our findings depicted that there is a similarity in reported and present research outcomes. These two top complexes were subjected to 100 ns MD simulations to confirm the stability and interactions between the target and compounds. The Root Mean Square Deviation was calculated for both complexes which showed that CYP51-Seocalcitol revealed different poses with no major difference and both complexes were stable. The Root Mean Square Fluctuation analyses for these two top complexes depicted that CYP51-Seocalcitol showed minor fluctuations in 220–270 residues while major fluctuations in 400 to 450 residues but with no effect because the residues in this range were not involved in interactions. The RMSF results revealed the overall stability of both complexes. The Principal Component analysis depicted a total variation of 50.8% for CYP51-Seocalcitol and 52.73% for CYP51-Isavuconazole. The MMGBSA showed a minor difference in ΔGtotal between the top two complexes as of about two points while both complexes were observed as stable with respect to binding energies. Overall, the simulation results were observed as compatible with Molecular docking outcomes. The binding affinity and interaction modes of Seocalcitol 70 are proximately similar to that of Isavuconazole in targeting the active sites of CYP51. The antifungal and anti-mucormycotic potential of Seocalcitol and most of the other vitamin D analogs especially Paricalcitol and Falecalcitriol, may be explicated by the fact that these sterol-like compounds may exert competitive inhibition in segregation of ergosterol component of the fungal membrane. The crucial role of azole anti-fungal group functions is to block lanosterol 14-alpha-demethylase, a fungus cellular membrane enzyme that converts lanosterol to ergosterol. Moreover, the anti-fungal activity of vitamin D analogs could be partly attributed to their hydrophobic and lipophilic properties, which enable them to penetrate the lipid bilayer, inducing alteration of permeability and subsequent cellular membrane leakage. 58
Conclusion
The CYP51 belongs to the CYP family. The involvement of CYP51 in fungal-specific ergosterol synthesis plays an important role in identifying the inhibitors by considering it as the target protein. C. albicans can cause mild to severe fungal infections in humans, while these infections can be inhibited by targeting the CYP51 protein in C. albicans. There are a large number of compounds and drugs available in the market and reported in the scientific research community, but there is still a need for developments in efficacy because of failure in therapeutics as well as in culture overwhelming azole resistance.
Vitamin D3 relevant compounds and its precursor 7-dehydrocholesterol have been extracted and detected in the leaves of many plants, particularly those belonging to Solanaceae species.
In this study, many in-silico analyses were done to examine the interaction of new inhibitors other than imidazoles, triazoles, tetrazoles, and many other traditional compounds or drugs. As well as we identified the most potent drug target sites (CYP51) against new compounds (synthetics, derivatives, and analogs of Vitamin D and D3) to inhibit antifungal infections. The 2D and 3D structure analysis was done to infer the secondary and tertiary structure insights. The molecular docking technique was utilized to inhibit the antifungal activity by targeting CYP51 with 15 Vit D, and D3 compounds and reported drugs in comparison. The top selected complexes from both categories were subjected to Molecular Dynamics simulation assays to confirm their stability. The in-silico analyses against drugs Fluconazole and Voriconazole depicted that the binding of these drugs is far away from the binding sites while the Isavuconazole binding is in the vicinity of the binding site and according to reported ones too. While most of the 15 Vit D, D3 compounds were observed in binding vicinity to some extent. So, these 15 compounds other than Alfacalcidol, Eldecalcitol, Inecalcitol, and Vitamin D3 can be further analyzed to develop a potent drug. Our findings suggest Vitamin D analog (Compound) and the prodrug (Isavuconazole) both showed the lowest binding affinities and are most related to binding cavities, with reported data as well as with each other, despite hydrogen bonds. Instability conformation, both complexes have shown the most similar results with minor differences. Wherefore, Seocalcitol could be further assessed to be adjacently utilized with the prodrug Isavuconazole to inhibit mucormycosis by targeting CYP51 while all these 11 Vit D, D3 compounds may lead to clinical trials after proper synthesis to increase the efficacy against antifungal activities specifically Seocalcitol can be chosen for first trial as it was further analyzed in this study. In the future, the other 10 compounds could also be subjects for further molecular dynamics analyses.
Footnotes
Author Contributions
Conceptualization: M.H.F.S. and G.M.A.; methodology M.H.F.S., G.M.A. and W.A.A.; formal analysis: M.H.F.S., G.M.A., M.M.A. and W.A.A.; writing—original draft preparation: M.H.F.S., A.A. and G.M.A.; writing—review and editing M.H.F.S., G.M.A., A.A. and W.A.A. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Deanship of Scientific Research, University of Hafr Al Batin for the research group project No. 0071-1443-S.
Ethical Approval
Ethical approval is not applicable to this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Institutional Review Board Statement
Not applicable.