
Abstract
Continuous scientific research is necessary to help in the discovery of new promising remedies for the treatment of COVID-19, caused by the SARS-CoV-2 virus. This current research was aimed at identifying potential novel inhibitors of the SARS-CoV-2 main protease, which represents one of the most important targets in the viral life cycle. Protein data bank file ID: 7JQ2 was used containing the co-crystallized inhibitor
Introduction
Evodia (Rutaceae) species are widely spread throughout Asia, Oceania, and eastern Africa, with twenty species and five variations described in the literature. 1 Among them is Evodia rutaecarpa Benth. (Wu-Chu-Yu), the dried unripe fruit of which is extensively used in traditional Chinese medicine for treating multiple gastrointestinal disorders such as headache, abdominal pain, and hemorrhage. 2 Moreover, it is even used as a cardiotonic due to its positive inotropic and chronotropic effects. 3 Phytochemical analysis of the extract revealed the alkaloids evodiamine (A), rutaecarpine (B), evodiamine (A), and rutaecarpine (B), which exhibited anti-inflammatory potential through either inhibition of cyclo-oxygenase 2 (COX-2) gene expressions or direct inhibition of COX-2, respectively. Furthermore, evodiamine (A) has been proven to prevent tumor cell proliferation, invasion, and metastasis while causing minimal harm to normal human peripheral blood cells. Its anticancer properties were observed through the stabilization of the complex formed between DNA and topoisomerase I, as well as the inhibition of heat shock protein (HSP) 27.4,5
The therapeutic potential of E rutaecarpa encourages its screening against other targets. This concept led us to elucidate its antiviral properties against several viral targets such as the recent corona virus (SARS-CoV-2) outbreak of 2019, which has claimed the lives of millions worldwide.
6
Inspired by the findings that suggest the effectiveness of favipiravir against SARS-CoV-2 RNA polymerase,7,8 previously reported research work has demonstrated the antiviral capabilities of several phytochemicals
9
such as evodiamine (

Methodology
Absorption, Distribution, Metabolism, Elimination, and Toxicity
The absorption, distribution, metabolism, elimination, and toxicity (ADMET) of the selected compounds were determined using Discovery studio 2.5 ADMET descriptor protocol. 13 All compounds were drawn using the built-in tool and then saved for various analyses.14,15
Molecular Docking
Molecular docking simulations were performed via MOE 2019 software. The receptor was downloaded from the protein data bank (PDB ID: 7JQ2). 16 The receptor and compounds were prepared and optimized using the standard structure optimization protocol of the software “QuickPrep,” and then compounds were compiled into a database. Afterward, both receptor and the ligands were prepared using the standard structure optimization protocol of the software. Energy minimization was performed using AMBER12: EHT force field and the active site was set to the pocket surrounding the co-crystalized ligands. The docking was performed using the MOE DOCKTITE Wizard-induced fit protocol using the GBVI/WSA dG scoring function. 17 Validation was achieved through re-docking of the co-crystalized ligand into the binding pocket and calculation of root mean square deviation (RMSD) between poses. Biovia DS Visualizer was used to analyze the docking results as well.18,19
Molecular Dynamics and Molecular Mechanics-Generalized Born Surface Area Calculations
The Schrödinger Desmond package was used for molecular dynamics simulations of free protein and complexes of protein with
The molecular mechanics-generalized born surface area (MM-GBSA) technique was used to compute the binding free energy of the protein–ligand complexes studied, which integrated MM force fields with a Generalized Born and Surface Area continuum solvation model using the Schrodinger Prime package.23,24
Results and Discussion
ADMET Study
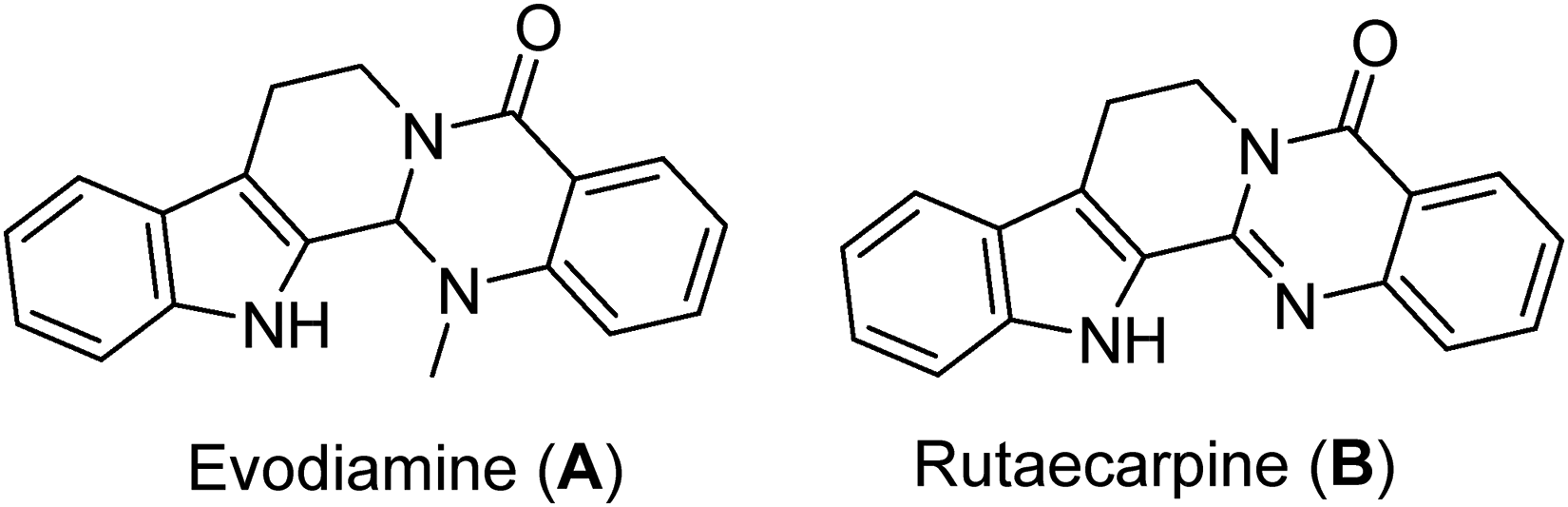
The predicted pharmacokinetic properties of the compounds showed that most of them possess good intestinal absorption, despite their poor solubility (Figure 1). Furthermore, the blood–brain barrier was permeable to nearly all of the compounds, except
(A) Predicted ADMET absorption chart. (B) Statistical analysis of ADMET properties.
ADME Properties for the Selected Natural Evodiamine Compounds.

S (Solubility): 0 (Extremely low), 1 (Very low), 2 (Low), 3 (Good), 4 (Optimal).
A (Absorption): 0 (Good), 1 (Moderate), 2 (Poor), 3 (Very poor).
BBB permeability: 0 (Very high), 1 (High), 2 (Medium), 3 (Low), 4 (Undefined).
Molecular Docking
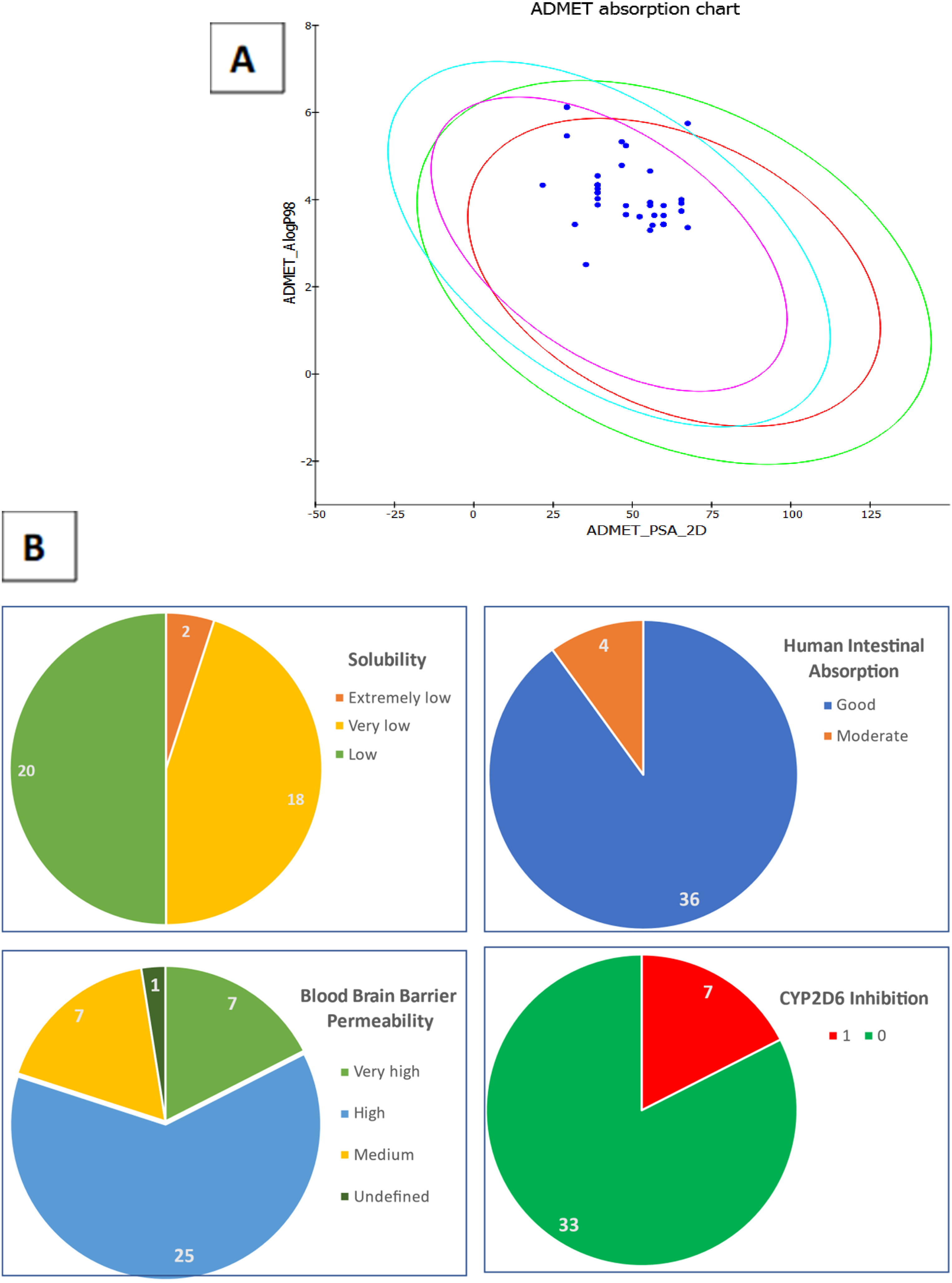
To explore the antiviral potential of evodiamine compounds, SARS-CoV-19 protease was selected and obtained from the protein data bank (PDB: 7JQ2).
16
Docking protocol validation was successful in redocking the co-crystallized inhibitor
Docking protocol validation: (co-crystallized pose = green and redocked pose = pink) inside SARS-CoV-2 main protease binding site.
Binding Affinities of Compounds Against SARS-CoV-2 in kcal/mol.

The

2D interactions of
The top five scoring molecules of our natural evodiamine library were

(A) 3D superimposition of
Interactions of the other top-scoring hits illustrated their binding to four common amino acid residues (His41, Cys145, Met49, and Met165) as

2D interactions of compounds
Consequently, the promising properties of the top-scoring compound
Molecular Dynamics
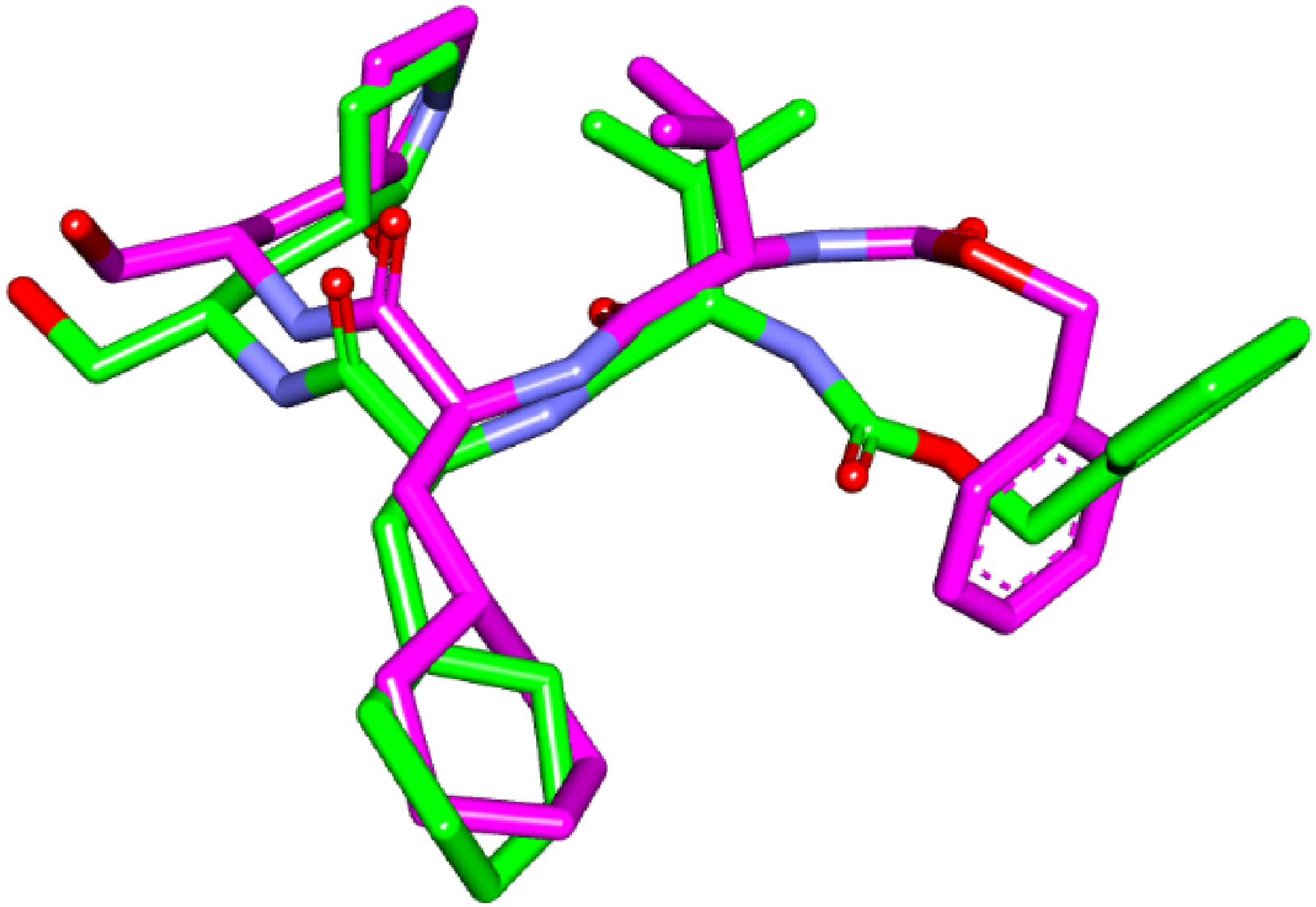
The molecular dynamic simulation was used for extensive analysis of the binding modes and stability under realistic physiological conditions. Using the Schrodinger Maestro suite, the protein with and without
The molecular dynamic simulation of the free protease shows relative homogeneity in behavior as demonstrated by RMSD's initial consistency around 2 Å for the first 40 ns, followed by a plateau at 2.60 Å (Figure 6). When
RMSD plots of protease in the absence and presence of inhibitors
Similarly, the same observation can be drawn when examining the RMSF values of the amino acid residues in the presence and absence of

RMSF plots of protease in the absence and presence of inhibitors
One of the most commonly used methods for calculating binding free energy is Molecular Mechanics Generalized-Born Surface Area (MM-GBSA). The lower a ligand-protein complex's projected binding free energy is, the more stable the complex is expected to be, and the higher the ligand's activity and potency (Table 3). Both complexes showed stable binding throughout the dynamic simulation.
MMGBSA of SARS-CoV-2 Protease in Complex with

Conclusion
The recent SARS-COV-2 (COVID-19) pandemic revealed the imbalance between drug research and health hazard outbreaks. We have tried to bridge the gap through investigation of the potential antiviral aspects taking advantage of modern computer-aided drug design such as virtual screening and molecular dynamics. Evodia (Rutaceae) species contain many compounds widely used in folk medicine for the treatment of various ailments. Among these compounds, research shed light on the antiviral properties of evodiamine (
Footnotes
Acknowledgement
The authors are grateful to the deanship of scientific research at Taif University for funding this project through Grant no (20211).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical Approval is not applicable to this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by deanship of scientific research at Taif University grant number (20211).
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.