
Abstract
Despite the contemporary advancements in the field of science and medicine, combating the coronavirus disease 2019 (COVID-19) is extremely challenging in many aspects as the virus keeps spreading and mutating rapidly. As there is no effective and conclusive drug therapy to date, it is crucial to explore plant-based natural compounds for their potential to inhibit SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). Recent research highly focuses on screening various phytochemicals to elucidate their anti-viral efficacy. However, very few studies were published investigating the anti-viral efficacy of ginsenosides. Hence, the main aim of this study was to investigate the inhibitory potential of the available 122 ginsenosides from Panax ginseng against SARS-CoV-2-related proteins using a molecular docking and molecular dynamics approach. The major bioactive compounds “ginsenosides” of P. ginseng were docked to six vital SAR-CoV-2 host entry-related proteins such as ACE2, Spike RBD, ACE2 and Spike RBD complex, Spike (pre-fused), Spike (post-fused), and HR domain, with lowest binding energies of −9.5 kcal/mol, −8.1 kcal/mol, −10.4 kcal/mol, −10.4 kcal/mol, −9.3 kcal/mol, and −8.2 kcal/mol, respectively. Almost all the ginsenosides have shown low binding energies and were found to be favourable for efficient docking and resultant inhibition of the viral proteins. However, ACE2 has shown the highest interaction capability. Hence, the top five ginsenosides with the highest binding energy with ACE2 were subjected to MD, post MD analysis, and MM/PBSA calculations. MD simulation results have shown higher stability, flexibility, and mobility of the selected compounds. Additionally, MM-PBSA also affirms the docking results. The results obtained from this study have provided highly potential candidates for developing natural inhibitors against COVID-19.
Introduction
Covid-19 is a disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This highly causative human coronavirus (CoV) was first reported in Wuhan, China, in December 2019, where pneumonia of unknown cause was detected. 1 These novel Coronaviruses (CoVs) belong to the family Coronaviridae, the order Nidovirales, and the genus Coronavirus, along with two other known CoVs SARS-CoV and the Middle East respiratory syndrome coronavirus (MERS-CoV). These three zoonotic viruses are the largest group of viruses discovered so far that cause more severe respiratory and gastrointestinal infections in humans than other known human CoVs, that only cause mild respiratory tract infections. 2
Computational biology plays a vital role in drug discovery, especially during epidemics. Advances in informatics and computational biology have increased productivity at many stages of the drug discovery pipeline. 3 Therefore, when the genome sequence of SARS-CoV-2 was first published, it allowed researchers to conclude that the new virus is closely related to SARS-CoV (82% sequence identity) and, to a lesser extent, to MERS-CoV. 4 This genomic sequencing is helping researchers and scientists search for novel and highly potent inhibitors more rapidly from natural compounds and repurposing already available FDA-approved drugs. Structural biologists have also solved more than 500 X-ray crystallography structures of SARS-CoV-2 proteins with good resolution. These are open to the scientific community through freely available databases. These data have encouraged many computational studies to predict the potential inhibitors from already known drugs. Several related articles have been published with a general protocol of extensive screening of drug databases via various molecular docking approaches, followed mainly by molecular dynamics simulations to increase the reliability of the docking results.5,6
Coronavirus is a single-stranded, positive sense RNA virus that does not have production capability. Hence, they infect and replicate in humans. Covid-19 drug discovery is based highly on two different targets, viral-based and host-based. 7 SARS-CoV-2 contains four major structural proteins: spike (S), membrane (M) and envelope (E) proteins, and nucleocapsid (N) protein. The S protein comprises the S1 subunit (which includes the N-terminal domain (NTD) and the receptor-binding domain (RBD) and the S2 subunit (which consists of fusion peptide (FP), connecting region (CR), heptad repeat 1 (HR1), heptad repeat 2 (HR2) and central helix (CH)). The SARS-CoV-2 S protein binds to its host receptor, the dimeric human angiotensin-converting enzyme 2 (hACE2), via the RBD and dissociates the S1 subunits. Cleavage at both S1–S2 and S2′ sites allows structural rearrangement of the S2 subunit required for virus-host membrane fusion. 8 Figure 1. illustrates the viral entry mechanism and targets of COVID-19.
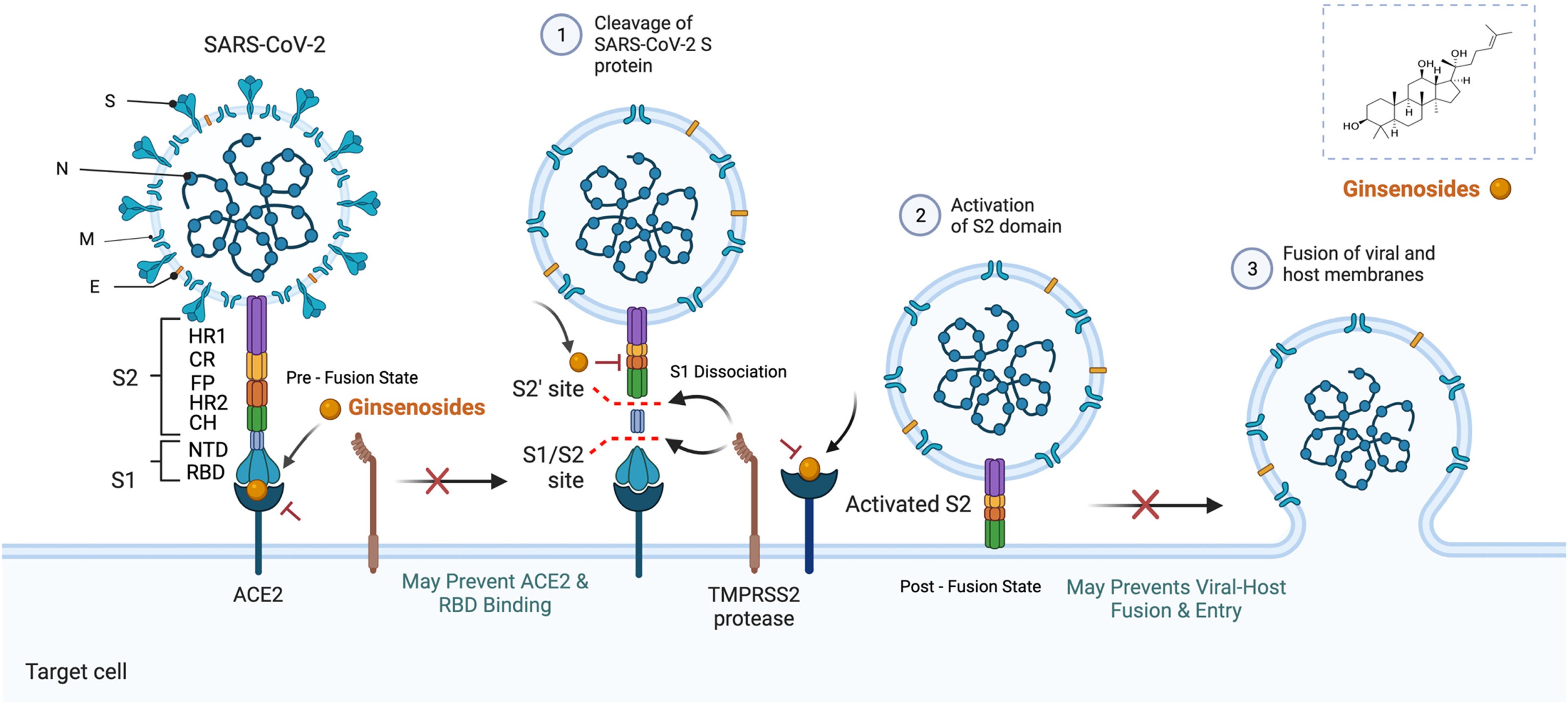
Illustration of the mechanism of action of ginsenosides against the main drug targets of COVID-19. Spike (S); Membrane (M); Envelope (E); Nucleocapsid (N); N-terminal domain (NTD); Receptor-binding domain (RBD); Fusion peptide (FP); Connecting region (CR); Heptad repeat 1 (HR1); Heptad repeat (HR2); Central helix (CH)).
Viruses mutate as they replicate and spread in a population. Viruses containing RNA as genetic material mutate much faster than viruses with DNA. For every replication of SARS-CoV-2, there is a high chance for the virus to change. Many mutations do not affect the virus's ability to spread or cause disease. On the contrary, if a mutation could affect the virus's ability to extend or cause disease that impacts public health, it will be categorized as a “variant of interest” or “variant of concern.” SARS-CoV-2 is one of the viruses that continuously evolve as changes in the genetic code caused by genetic mutations or viral recombination occurs during genome replication. The number of SARS-CoV-2 variants and mutations to date is as follows: Alpha (B.1.1.7 and Q lineages), Beta (B.1.351 and descendent lineages), Gamma (P.1 and descendent lineages), Delta (B.1.617.2 and AY lineages), Epsilon (B.1.427 and B.1.429), Eta (B.1.525), Iota (B.1.526), Kappa (B.1.617.1), 1.617.3, Mu (B.1.621, B.1.621.1), Omicron (B.1.1.529, BA.1, BA.1.1, BA.2, BA.3, BA.4 and BA.5 lineages), and Zeta (P.2).
The CDC categorizes Delta and Omicron variants as the “variants of concern.” In 2020, Delta was first identified in India, spreading to over 179 countries by November 22nd, 2021, becoming the dominant strain worldwide. On April 14th, 2022, the U.S government SARS-CoV-2 Interagency Group (SIG) downgraded Delta from a “Variant of Concern” to a “Variant Being Monitored. 9 ” On November 30th, 2021, SIG classified Omicron as a Variant of Concern (VOC). Since then, the death rates have been higher from Omicron than Delta, with the Omicron variant accounting for 99.9% of all COVID-19 cases in the United States. 10 Reportedly, the Omicron variant is associated with high transmissibility leading to high infectivity and increased reinfection rates. Compared with Delta, the Omicron variant demonstrates two main and ‘exotic’ molecular features: a combination of deletions and one insertion in S1/RBD and S2 spike regions, respectively. Interestingly, a fragment of highly conserved point substitutions demonstrates a remarkable penetration. 11 The Delta variant had around ten mutations in the spike protein. In contrast, the Omicron had approximately thirty-two (15 of which occur in the RBD, and three small deletions and one minor insertion).12,13 Currently, Omicron is the “Variant of Concern,” with around 6,487,955 deaths and 601,454,808 cases worldwide. 14 Moreover, the cases are expected to increase as new variants emerge. Hence, there is a crucial need for new drugs and therapeutics to combat the current and emerging variants.
Currently, there is no available drug therapy to treat COVID-19. Therefore, drugs are needed to inhibit the vast and rapid spread of SARS-CoV-2. Natural resources are an excellent source of chemical compounds applicable to various ailments. Since the beginning of the pandemic, numerous in silico studies highly focus on screening and repurposing potential inhibitors from natural products for their anti-viral efficacy against SARS-CoV-2.15–23 Natural products such as plant metabolites comprise a vast and diverse source of bioactive compounds, some of which are supported by thousands of years of traditional medicine. Because of their varied therapeutic applications, one among them is “ginsenosides,” major bioactive components of Panax ginseng, family Araliaceae. There are 15 species and seven subspecies of Panax. Among the species of Panax, only four have been studied widely: P. ginseng (Korean ginseng), P. notoginseng (Chinese ginseng), P. quinquefolius (American ginseng), and P. japonicus (Japanese ginseng). 16 The various ginseng processing methods produce a range of ginsenoside compositions with diverse pharmacological properties, which are reported elsewhere. 17 Depending on the differences in their chemical compositions and configurations, ginsenosides are classified into three types: panaxadiol, panaxatriol, and oleanolic acid. The significant ginsenosides isolated from ginseng (including Rb1, Rc, Rd, Re, and Rg1) typically account for more than 70% of total ginsenoside content. Ginsenosides are often used as quality indicators for assessing ginseng products. 18 More than 289 saponins have been reported from eleven different Panax species. In addition, at least 123 ginsenosides have been identified in Panax species, including both naturally occurring compounds and those produced from steaming and biotransformation. Although most of the ginsenosides have a rigid four-trans-ring steroid skeleton, they produce multiple pharmacological and biological effects that are different from one to another due to minor variations in (1) Type of sapogenin; (2) number, type, and site of glycosyl units; and (3) modification of C17 side-chain. 19
Ginsenosides interact with various signalling pathways and effect changes at the transcription level, exhibiting different benefits. 20 Ginsenosides are known for their antioxidant, anti-inflammatory, anti-microbial, anti-cardiovascular, anti-obesity, and anti-diabetic activities. In 2016, a review focusing on the anti-viral efficacy of Korean red ginseng was published, compiling the in vivo and in vitro evidence for various ginsenosides, such as Re, Rf, Rg2, Rb1, Rb2, Rc, Rd, Rh1, Rh2, and Rg3. 21 Recently, a few studies have been published focusing on the preventive efficacy of ginseng on SARS-CoV-2 and its related symptoms.22,23 A recent study has investigated the anti-viral efficacy of the fermented black ginseng extract, which inhibits the replication of this virus strain in the SARS-CoV-2 infected Vero E6 cell, but also reduces the number of viral RNA copies in the extracellular environment. 24 Additionally, ginsenosides are reported to mimic the action of steroids in binding and explicating similar activities via binding to glucocorticoid receptors. Ginsenoside Rg1 is a natural plant compound with structural similarity to classical glucocorticoids. Ginsenosides Rg1 and Re are known functional ligands of the glucocorticoid receptor (GR). 25 In 2018, a study analyzed the glucocorticoid-like activity of Protopanaxadiol (PPD) and Protopanaxatriol (PPD) with dexamethasone as control and showed that they could be considered potent and selective GR agonists. In addition, the structural similarities of PPD and PPT with cortisol and the synthetic corticosteroid dexamethasone (DEX) have been shown, and its binding potential to the GR ligand binding domain (LBD) has been examined by applying induced-fit docking analysis, which showed better binding predictions. 26 The ability of ginsenosides to regulate the non-genomic effect of GR has also been examined. 27 Currently, some clinical studies have reported using a few drugs in severe cases, such as the steroid dexamethasone, which improved survival in cases of COVID-19. 28 In addition, another study revealed that compassionate use of remdesivir produced clinical improvement in 68% of hospitalized patients with severe cases of COVID-19. 29 Dexamethasone inhibits the entrance of SARS-CoV-2 spike pseudotyped virus into the cell by binding to ACE2. 30 Dexamethasone is a promising drug, but with many side effects. Hence, an alternative and potential natural inhibitor against the related viral proteins is highly needed.
This study aimed to investigate the efficacy of ginsenosides in preventing and treating SARS-CoV-2. Figure 1 illustrates ginsenosides’ mechanism of action as drug targets. In the present study, a library of 122 ginsenosides and seven known drug controls (Dexamethasone, Remdesivir, Chloroquine, Arbidol, Disulfiram, Indinavir, Comostat) have been subjected to virtual screening using molecular docking against six SARS-CoV-2 viruses, as well as host entry proteins (ACE2, Spike RBD, ACE2 and Spike RBD complex, Spike (pre-fused), Spike (post-fused), HR domain). The top five ranked ligands for each protein have been visually analyzed, and their interactions reported. Finally, the top-ranked protein-ligand complexes for the protein with the best docking results have been verified using molecular dynamics simulation studies, and their RMSD, RMSF, Radius of Gyration, the number of hydrogen bonds, and MM-PBSA calculations were analysed.
Results and Discussion
Ginsenosides are valuable compounds with diverse pharmacological properties that can aid in managing several diseases. However, few studies have evaluated and published their anti-viral (COVID-19) efficacy.22,23 Therefore, this study evaluated the anti-viral effectiveness of ginsenosides (122) via computational approaches, as they are the principle and fundamental investigation methodology that has been used widely in recent times. Computational techniques are vital to discover novel and potential small molecules in drug discovery and development as they are cost-, time-, and labor-effective. ACE2 plays a unique role in regulating viral attachment and entry. Once ACE2 fuses with RBD, the s1 subunit of the spike protein is dissociated, and the s2 subunit will further undergo structural rearrangement. Therefore, it will enter the host cell to replicate (Figure 1). Hence, it is essential to inhibit the post-fused s2 subunit, which will avoid fusion and thus can prevent further infection and replication. In addition, the viral fusion mechanism of SARS-CoV-2 mainly depends on the interaction of heptad repeat 1 and 2 (HR1 and HR2) regions in the spike protein. Therefore, Heptad repeat 2 (HR2) domains involved in the fusion of spike glycoprotein trimers (viral membrane fusion) were considered a significant target.
Firstly, we screened the ginsenosides and control drugs for their inhibitory activity against six SARS-CoV-2 viruses, as well as host entry proteins (ACE2, Spike RBD, ACE2 and Spike RBD complex, Spike (pre-fused), Spike (post-fused), HR domain) through molecular docking techniques. The in-silico evidence from our study suggests that all the docked protein targets have greater binding affinity with ginsenosides. Furthermore, the MD simulation study showed the time-dependent stability of ligand-receptor complexes for a period of 100 ns. Therefore, as a final step, we employed MM-PBSA free binding energy calculations to verify further the MDS results, which also showed a promising result approving the prior validations. ACE2 and all other targets in this study have also demonstrated significant results confirming the anti-viral efficacy of ginsenosides. Hence all the above facts prove that natural compounds like ginsenosides can be considered effective inhibitors of the principal viral and host entry targets of SARS-CoV-2 and thus can deliver effective treatment to the highly infectious viral disease Covid-19. All the results from this study are in detail as follows:
Molecular Docking Study
We performed a molecular docking study against the six protein targets of SARS- Cov-2 using Auto Dock Vina. As a result, the best five top-scored ligands, four control drug inhibitors, and the inhibitor separated from the native protein were analyzed by post docking analysis. The interactive sites and the interacting amino acids involved are represented in Figure 2 and Figure S1–5. The figures represent the 3D interaction of the top-ranked ligand. In addition, the 2D interaction diagram for the other four top-ranked ligands and the control drugs for each of the selected target proteins has been given along for comparative analysis. As a result, most of the ginsenosides have shown significant binding energy with all the target proteins used in this study (Supplementary File).
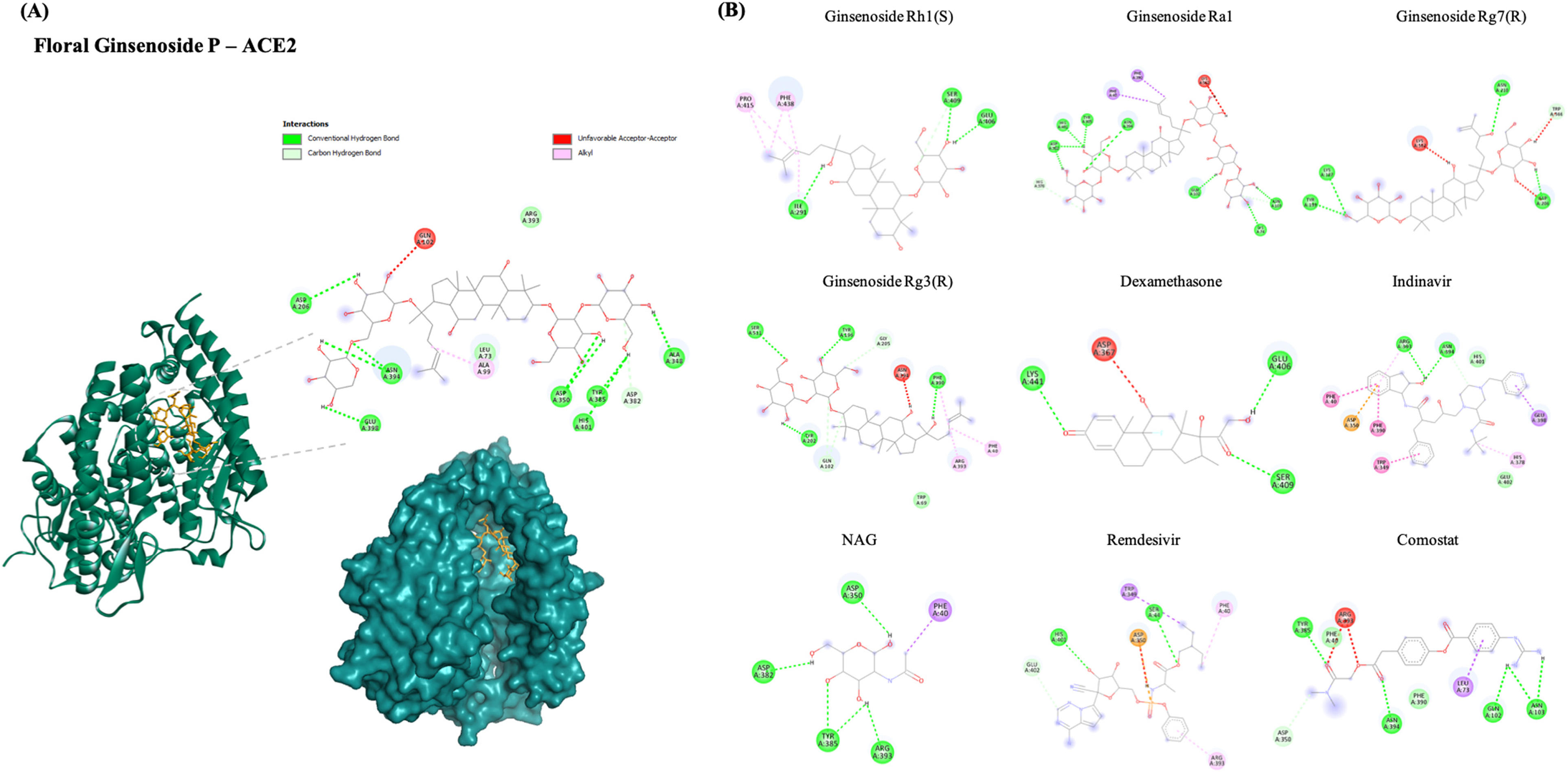
(A) 2D and 3D docking interactions of the best binding ginsenoside (Floral Ginsenoside P) with ACE2 (PDB ID: 6M0J). (B) Docking interactions of top ranked ginsenosides (Ginsenoside Rh1(S), Ginsenoside Ra1, Ginsenoside Rg7(R), Ginsenoside Rg3(R)) and control drug inhibitors (Dexamethasone, Indinavir, NAG, Remdesivir, Comostat with ACE2).
Interaction with Human ACE-2
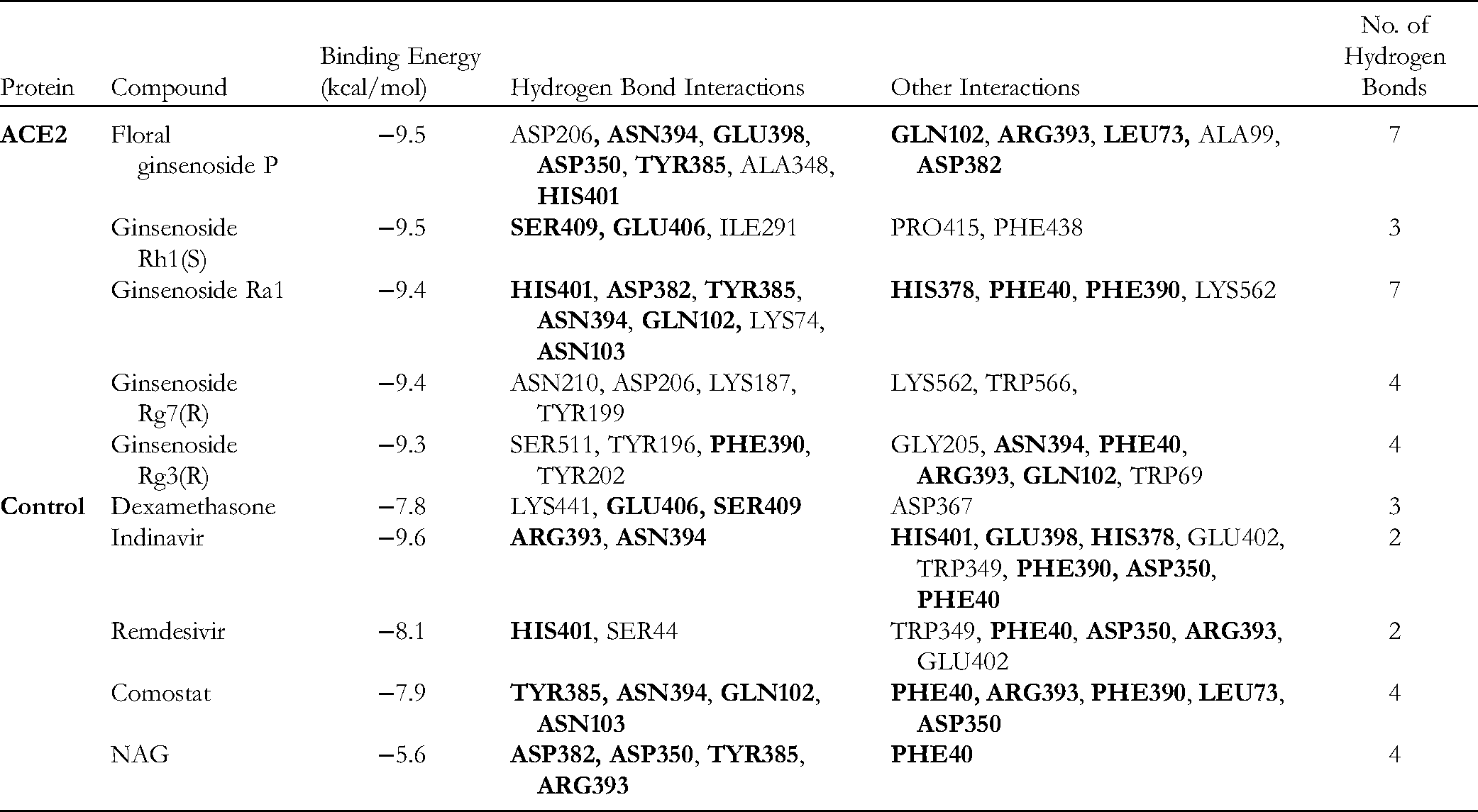
Angiotensin-converting enzyme 2 (ACE2), the functional receptor of SARS-CoV-2, plays a crucial role in the pathogenesis of COVID-19, as it provides viral entry into human cells.31,32 Figure 2 shows the interactions between the best ligand for Floral Ginsenoside P with the lowest binding energy −9.5 kcal/mol against the human ACE2 protein. Floral Ginsenoside P formed 7-H bonds with ASP-206, ASN-394, GLU-398, ASP-350, TYR-385, ALA-348, and HIS-401 residues, and five hydrophobic interactions with GLN-102, ARG-393, LEU-73, ALA-99, and ASP-382. The binding energies of ginsenosides with ACE2 were more significant than most of the control molecules utilized in this study, indicating that ginsenosides have higher binding activity. The binding site of Floral Ginsenoside P is close to that of the control molecule NAG, which was separated from the native PDB structure. It also shares two similar 2-H bonds, the same as NAG (ASP350, TYR385).
The binding energies of Ginsenoside Rh1(S), Ginsenoside Ra1, Ginsenoside Rg7(R), and Ginsenoside Rg3(R) to ACE2 were −9.5 kcal/mol, −9.4 kcal/mol, −9.4 kcal/mol, and −9.3 kcal/mol, respectively (Table 1). As a result, the higher binding energy and similar H-bond and other types of interaction to controls prove that Ginsenosides have strong interactions with the main host-based target ACE2 of the new coronavirus. This study shows that ginsenosides might be promising ACE2 inhibitors. Hence, the top five ginsenosides with ACE2 complexes have been further utilized for molecular dynamics simulation to validate the docking scores and other properties, such as the stability and flexibility of the docked complex.
Interaction of Top Five Ginsenosides with Amino Acid Residue of ACE2 (6M0J).
Interaction with Spike RBD
RBD is a crucial functional component within the S1 subunit of the spike glycoprotein responsible for the binding of SARS-CoV-2 to ACE2. Hence, the inhibitory efficacy of ginsenosides on Spike RBD was evaluated in this study, as it is one of the essential drug targets of COVID-19. Figure S1 shows the interactions formed between the best ligand (20S)-Ginsenoside Rg2 with the lowest binding energy of −8.1 kcal/mol against the RBD of Spike glycoprotein. (20S)-Ginsenoside Rg2 forms 3-H bonds with the residues ASP-206, ASN-394, GLU-398, ASP-350, TYR-385, ALA-348, and HIS-401.Other top-ranked ginsenosides are Ginsenoside Ro, Ginsenoside Ra1, Ginsenoside Compound Y, and Ginsenoside Rg3(S) with −8.1 kcal/mol, −8.0 kcal/mol, −7.9 kcal/mol, and −7.9 kcal/mol of binding energy to RBD, respectively. Interestingly, the binding energies of ginsenosides to the active site are even better than those of the control molecules (Table S1).
Interaction with Spike RBD with ACE-2 Complex
Ginsenoside Ra1 is one of two ginsenosides that showed the lowest binding energy (−10.4 kcal/mol) against one of the proteins that target RBD of spike glycoprotein and ACE-2 complex, as shown in Figure S2. Ginsenoside Ra1 formed 8-H bonds with GLN409, HIS34, GLN493, SER494, GLU37, TYR505, THR27, and TYR473 residues. In addition, it also showed hydrophobic interactions with LYS417 and TYR495 residues of the target protein. The binding energy of other top-ranked ligands, such as Floral Ginsenoside P, Floral Ginsenoside O, Ginsenoside Rg2(R), and Ginsenoside Ki, was −9.5 kcal/mol, −9.4 kcal/mol, −9.4 kcal/mol, and −9.3 kcal/mol, respectively, as shown in Table S2.
Interaction with Pre-Fused State of S Protein
Floral Ginsenoside E formed the lowest binding energy (−10.4 kcal/mol) with the spike glycoprotein's pre-fused state, as shown in Figure S3. Further, it formed 5-H bonds with residues LYS1045, ASP1041, LYS1028, THR1027, and AGR1039. In addition, Floral Ginsenoside E built other hydrophobic interactions with the AGR1039 residue. Also, one amino acid residue, ASP1041, is similar to that of the control inhibitor NAG (ligand obtained from the native PDB structure of the protein). The binding energy of the subsequent top-ranked ligands, such as Ginsenoside Rh1(R), Floral Ginsenoside C, Ginsenoside Rh1(S), and Ginsenoside Rg3(S), was −9.7 kcal/mol, −9.6 kcal/mol, −9.4 kcal/mol, and −9.4 kcal/mol, respectively (Table S3).
Interaction with Post-Fused State of S Protein
Figure S4 shows the interactions between the best ligand, Ginsenoside Rg5, with the lowest binding energy (−9.3 kcal/mol) against the post-fusion state of spike glycoprotein. Ginsenoside Re formed 1-H bond with the residue ASN1194 and three hydrophobic interactions with ASN1194, ASN925, and LEU1197 residues. The binding energy of the following top-ranked ligands, Floral Ginsenoside I, Ginsenoside Rg3(R), Ginsenoside Rh9, and Ginsenoside Ro, to the post-fused state of S protein was −9.3 kcal/mol, −9.2 kcal/mol, −9.1 kcal/mol, and −9.1 kcal/mol, respectively (Table S4).
Interaction with HR Domain
The viral fusion mechanism of SARS-CoV-2 mainly depends on the interaction of heptad repeat 1 and 2 (HR1 and HR2) regions in the spike protein. Therefore, Heptad repeat 2 (HR2) domains involved in the fusion of spike glycoprotein trimers (viral membrane fusion) were considered a significant target. Figure S5 shows the interactions between the best ligand Ginsenoside Re with the lowest binding energy (−8.2 kcal/mol) against the HR domain of spike glycoprotein. In addition, Ginsenoside Re forms 2-H bonds with residues ARG18 and GLU21 and three hydrophobic interactions with the ALA23, LYS24, and ARG18 (Table S5). The binding site of Ginsenoside Re is close to that of the control drugs, Dexamethasone and Comostat, also sharing 2-H bonds with NAG (ARG18, GLU21).
Molecular Dynamic Simulation Studies
For further investigation, the molecular docking results of the top four docked ligands with the crystal protease complex ACE-2 (PDB ID: 6M0J) have been verified using molecular dynamics simulation (100 nanoseconds). From the MD simulation trajectory, we analyzed the root mean square deviation (RMSD), root mean square fluctuation (RMSF), the radius of gyration (Rg), and the number of hydrogen bonds. These calculations were analyzed to study the receptor-ligand conformational properties, such as stability, flexibility, and H-bond interactions. From the docking validation, Floral Ginsenoside P with ACE2 had the highest docking score ( − 9.5 kcal/mol) and multiple amino acid residue interactions. Thus, the best-ranked ligand of ACE2, along with subsequent ginsenosides and controls, was simulated for 100 nanoseconds under the NTP ensemble. As a result, we performed RMSD and RMSF calculations to analyze the trajectory generated after the MD simulation. The RMSD (root mean square deviation) values show that the position did not change much once the ligands stabilized (Figure 3 (A)). In addition, all the ligand complexes showed almost the exact positional change and stability. Later, the RMSF (root mean square fluctuation) values were calculated to average the fluctuations of the positions of each residue to check the flexibility and mobility of a complex during the simulation (Figure 3 (B)). Then, the radius of gyration was used to study the overall effect of the ligands on the protein. First, the radius of gyration was calculated for each complex, as shown in Figure 4 (A).

(A) RMSD of ginsenosides in complex with SARS-Cov-2 ACE2 as a function of MD simulation time. (B) RMSF of ginsenosides in complex with SARS-Cov-2 ACE2 as a function of MD simulation time (100 nanoseconds).

(A) Radius of gyration plots of molecular dynamics (MD) simulation of ACE2 receptor-ginsenoside complexes. (B) Line plots of ligand-protein H bonds for ACE2 and ginsenosides.
Furthermore, the number of hydrogen bonds has been calculated and shown in Figure 4 (B). Overall, it was observed that Ginsenoside Ra1 formed more hydrogen bonds compared to other ligands. These MDS results are similar yet slightly different from the docking results. As a result, the MD simulation study reveals that ginsenosides can successfully interact with ACE-2 to form a stable complex. Finally, we performed MM-PBSA analysis to calculate the thermodynamics parameters of the complex, such as binding free/van der Waals/electrostatic/polar solvation energies (ΔEbinding; Evdw; Eelec; ΔEpolar, respectively) at the molecular level. Table 2 displays the calculation result, indicating that each complex's BFE value is greater than the control complex value. For example, the highest level of BFE (−72.71 kcal/mol) was the Ginsenoside Ra1-ACE2 complex.
Calculated Binding Free Energy (MMPBSA) for ACE2 Complexes Post MD Simulation.

Conclusion
In conclusion, ginsenosides from Panax ginseng are a potential natural inhibitor of SARS-CoV-2 related proteins. To study further the application of these natural compounds in the treatment and prevention of COVID-19, the docking scores and interaction sites of ginsenosides with the targets from this study are valuable. Almost all the ginsenosides showed low binding energy with all the selected receptors of SARS-CoV-2. Among all the compounds, Floral Ginsenoside P, E, C, I, and O, Ginsenoside Ra1, Rh1, Rg7, Ro, Rg3, Rg2, Ki, Rh9, and Re showed better antiviral activity against all the selected targets. They showed a high affinity towards the SARS-CoV-2: ACE2 interface and interacted with several hotspot residues through hydrophobic and hydrophilic bonding. Notably, the ginsenosides also showed better docking scores than the drug controls in this study, sharing many similar amino acid residues. In addition, MD simulation studies and MM-PBSA calculations also confirmed the stability of the ginsenoside and target protein complexes, which were analyzed based on their RMSD, RMSF, the radius of gyration, H-bond, and free binding energy values. These results suggest the potential inhibitory activity of ginsenosides in the treatment of COVID-19. Therefore, using ginsenosides in preference to synthetic drugs such as dexamethasone is an exciting treatment option. The results presented in the study will lead to evaluating the broad-spectrum antiviral activity of the ginsenosides in treating SARS-CoV-2. Eventually, in vitro, animal experiments, and accurate clinical trials are needed to confirm these compounds’ potential preventive and treatment effects.
Materials and Methods
Ligand Selection and Preparation
For the present study, we selected 122 ginsenosides belonging to P. ginseng. According to previous reports, a few of these have been shown to possess anti-viral efficacy. Hence this study intended to screen all the available ginsenosides. The chemical structures were obtained from PubChem 33 and for the ones not found in PubChem, the structures were sketched manually in ACD ChemSketch 34 and then converted into 3D structures. All the compounds and reference compounds were converted to PDB format using Open babel for further analysis. 35 The ligand molecules were further processed and converted to the required pdbqt format using Autodock tools. 36
Protein Targets Selection and Preparation
The drug targets of SARS CoV-2 were identified based on the literature survey, database search, and knowledge of their role in pathogenicity, especially in mediating host cells. The significant targets involved in the host entry virulence mechanism of SARS-CoV-2 are spike glycoproteins and ACE2. Six probable drug targets, namely ACE2(PDB ID:6MOJ), 37 Spike RBD (PDB ID:6XC4), 38 ACE2 and Spike RBD complex (PDB ID:6MOJ), 37 pre-fusion spike glycoprotein with a single receptor-binding domain (PDB ID:6VSB), 39 the post-fusion core of S2 subunit (PDB ID:6LXT), 40 and HR2 domain of S2 subunit (PDB ID: 6LVN), 41 were identified as potential molecular targets. The three-dimensional (3D) structures of all the selected proteins are available in their native forms and were downloaded from the RCSB PDB database 42 in PDB format. The structure preparation consisted of several steps, such as deleting all water molecules and inhibitors (ligands), checking, and repairing the missing atoms, and adding hydrogens and required charges using Autodock tools. The modified file was then saved in the required format (pdbqt) for docking analysis.
Molecular Docking Studies
Autodock Vina provides better ligand-protein binding poses and accuracy than AutoDock 4.2. Hence, we used AutoDock Vina 43 for this study's docking experiments to dock the selected 122 ginsenosides (122) and seven control drugs against the six selected protein targets. The grid box was created with the size of 70 Å × 70 Å × 70 Å and the other parameters were set as default. Compounds were then ranked based on their docking scores that represent binding energies. Finally, we have utilized the academic version of PyMOL 44 and BIOVIA Discovery Studio Visualizer (BIOVIA, Dassault Systèmes) 45 to visualize the ligand interactions with the active sites of the receptors.
Molecular Dynamics (MD) Simulation Studies
Molecular dynamics simulation for the best-ranked compounds (Floral Ginsenoside P, Ginsenoside Rh1, Ginsenoside Rg3, Ginsenoside Ra1) with ACE2 protein was performed for 100 ns using GROMACS 2020.1 software. 46 We utilized Swiss PARAM to produce the topologies of drugs. 47 The receptor-ligand complexes were placed in a periodic cubic solvated box, and an explicit SPC water model solvated the cell. Appropriate counter ions such as Na or Cl were used to neutralize the system. Energy minimization and equilibration for all systems were executed using NVT and NPT ensembles. Finally, we performed the MD simulation for 100 ns time steps. Next, we used the output data such as RMSD, RMSF, Rg, and the number of hydrogen bonds to analyse MD trajectories using the GROMACS utilities. Lastly, we employed the gmx_MMPBSA 48 package for free energy calculations based on the single trajectory of GROMACS with an appropriate force field. This tool allows free energy calculations using MM/PBSA or GBSA (Molecular Mechanics/ Poisson-Boltzmann or Generalized Born Surface Area) methods with an implicit solvent model.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221134331 - Supplemental material for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets
Supplemental material, sj-docx-1-npx-10.1177_1934578X221134331 for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets by Pang Shifeng, Vinothini Boopathi, Mohanapriya Murugesan, Ramya Mathiyalagan, JongChan Ahn, Chen Xiaolin, Dong-Uk Yang, Gi-Young Kwak, Byoung Man Kong, Deok-Chun Yang and Se Chan Kang, Zhang Hao in Natural Product Communications
Supplemental Material
sj-docx-2-npx-10.1177_1934578X221134331 - Supplemental material for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets
Supplemental material, sj-docx-2-npx-10.1177_1934578X221134331 for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets by Pang Shifeng, Vinothini Boopathi, Mohanapriya Murugesan, Ramya Mathiyalagan, JongChan Ahn, Chen Xiaolin, Dong-Uk Yang, Gi-Young Kwak, Byoung Man Kong, Deok-Chun Yang and Se Chan Kang, Zhang Hao in Natural Product Communications
Supplemental Material
sj-xlsx-3-npx-10.1177_1934578X221134331 - Supplemental material for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets
Supplemental material, sj-xlsx-3-npx-10.1177_1934578X221134331 for Molecular Docking and Dynamics Simulation Studies of Ginsenosides with SARS-CoV-2 Host and Viral Entry Protein Targets by Pang Shifeng, Vinothini Boopathi, Mohanapriya Murugesan, Ramya Mathiyalagan, JongChan Ahn, Chen Xiaolin, Dong-Uk Yang, Gi-Young Kwak, Byoung Man Kong, Deok-Chun Yang and Se Chan Kang, Zhang Hao in Natural Product Communications
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through Agri-Food Export Business Model Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (Project No: 320104-03); the Science and technology development project of Jilin Province (NO. 20210509022RQ); and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant no: NRF- 2020R1I1A1A01070867).
Author Contributions
Conceptualization: V.B. and P.S.; writing—original draft preparation: V.B.; Formal analysis: V.B., P.S., M.M., J.C.A. and C.X.; Funding acquisition and supervision: D.C.Y., Z.H., S.C.K., and R.M.; Resources: D.U.Y., G.Y.K., B.M.K. and D.C.Y. All authors have read and agreed to the published version of the manuscript.
Ethical Approval
Not applicable.
Informed Consent
Not applicable.
Trial Registration
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.