
Abstract
Overexpression of programed death-ligand 1 (PD-L1) is associated with poor prognosis in leukemia. Moreover, antitumor pharmaceuticals have been shown to induce immunoresistance, leading to reduced efficacy. Previous studies have indicated that arsenic trioxide (ATO) promotes immune evasion by inducing PD-L1 expression in solid tumors; however, little is known about its role in leukemia. A proportion of patients with acute promyelocytic leukemia were resistant to ATO therapy. Thus, this study aimed to investigate the effect of ATO on the expression of PD-L1 in leukemia cells and the underlying mechanism mediated through the nuclear factor erythroid 2 related factor (NRF2) protein. Brusatol, extracted from Brucea javanica, was selected as a unique NRF2 inhibitor, and we evaluated the possibility of using a regimen combining ATO/Brusatol in leukemia therapy. Promyelocytic NB4 and lymphocytic Jurkat cells were treated with ATO and brusatol either alone or in combination. We found that ATO significantly upregulated the expression of PD-L1 in NB4 and Jurkat cells at both the protein and mRNA levels compared with its expression in the untreated cell group. Mechanistically, ATO increased nuclear NRF2 expression and the extent of NRF2 binding to the PD-L1 promoter. Pharmacological inhibition of NRF2 by brusatol significantly blocked this effect, thereby reducing ATO-induced PD-L1 expression. In addition, the combination of brusatol and ATO showed stronger cytotoxicity than ATO alone indicated by cell counting kit-8 assay. Therefore, brusatol may further enhance the antileukemia effect of ATO not only by inhibiting ATO-induced PD-L1 expression but also by enhancing ATO-induced cytotoxicity. Our study provides a rationale for the clinical application of ATO/brusatol combination therapy.
Introduction
Recent investigations have suggested that a host-dependent immune response is indispensable for sustained tumor clearance. 1 Unfortunately, leukemia blasts are endowed with immune evasion mechanisms, indicating that they play roles in the immunosuppressive microenvironment which inhibits antileukemia immune reactions. One of the immune evasion mechanisms involves an increased abundance of programed death-ligand 1 (PD-L1) in tumor cells, which leads to increased exhaustion and apoptosis of T cells through PD-L1/PD-1 ligation, thus impairing the T-cell-mediated antitumor immune response. 2 Previous studies validated that PD-L1 is expressed in human leukemic blasts and that its expression is significantly higher in patients with relapsed disease than in patients with de novo leukemia. 3 Additionally, Chen et al. 4 reported that overexpression of PD-L1 in leukemic blasts is associated with poor overall survival of patients with acute myeloid leukemia (AML). Surprisingly, many studies have indicated that antitumor agents may aggravate immune evasion via their induction of PD-L1 expression. For example, chemopreventive agents such as paclitaxel, etoposide and 5-fluorouracil can induce PD-L1 upregulation in human breast cancer cells, and overexpressed PD-L1 increases T-cell apoptosis.5,6 This potential link between antitumor pharmaceuticals and immunoresistance has also been reported in leukemia. A previous study revealed that all-trans retinoic acid can enhance IFN-γ-induced PD-L1 upregulation through the induction of myeloid maturation. 7 Additionally, a clinical study reported that PD-L1 expression was upregulated in patients with AML during epigenetic therapy and that the patients with PD-L1 upregulation were more resistant to the therapy. 8 A recent study showed that arsenic trioxide (ATO) induced upregulation of PD-L1 in oral squamous carcinoma cells, which significantly reduced its anticancer activity. 9 However, little is known about the role played by PD-L1 in leukemia. Previous studies have demonstrated that a proportion of acute promyelocytic leukemia (APL) patients are resistant to ATO therapy. 10 Therefore, our study investigated the effect of ATO on PD-L1 expression in leukemia cells and its possible mechanisms due to the importance of ATO to leukemia therapy.
Nuclear factor erythroid 2 related factor (NRF2) is a key transcription factor of antioxidant proteins. Under oxidative stress, NRF2 is translocated to the nucleus, where it binds to the antioxidant response element of targeted genes, thus increasing the expression of a series of cytoprotective genes. 11 However, constitutive activation of NRF2 has been found in cancers, including leukemia, and hyperactive NRF2 can activate downstream effectors that promote not only metabolism, proliferation and survival of cancer, but also the development of chemoresistance in solid tumors and leukemia.12,13 NRF2 inhibition can effectively augment the antitumor effects of various drugs, including 5-fluorouracil and doxorubicin.14,15 In addition, recent studies have revealed that NRF2 also plays an important role in immune evasion in cancer. A study of lung cancer has reported that patients overexpressing NRF2 were resistant to PD-L1 blockade therapy, suggesting a potential link between NRF2 and the PD-L1/PD-1 axis. 12 Another study on melanoma found that the NRF2 pathway directly increased the expression of PD-L1, while NRF2 inhibition significantly reduced PD-L1 expression and increased the infiltration of T cells into tumor sties. 16 However, no studies have clarified the impact of NRF2 on PD-L1 expression in leukemia cells.
Brusatol, extracted from Brucea javanica, is a unique NRF2 inhibitor, that has been recognized as a potent antitumor drug. In particular, brusatol is a sensitizer when used in combination with other traditional chemotherapies. 17 However, there has been little investigation into its effects on PD-L1 expression.
In this study, we investigated the effect of ATO on the expression of PD-L1 in leukemia cells and the mechanism by which NRF2 mediates this process. The present study revealed that ATO increased the expression of PD-L1 mediated via NRF2 in leukemia cells and that pharmacological inhibition of NRF2 by brusatol decreased ATO-induced PD-L1 upregulation. In addition, brusatol enhanced ATO-induced cytotoxicity in leukemia cells. ATO combined with brusatol is expected to be an effective therapeutic regimen for leukemia.
Results
ATO-induced PD-L1 Upregulation in Leukemia Cells
Previous studies reported that ATO induced upregulation of PD-L1 in solid tumor cells. 9 To verify its effect on leukemia cells, ATO was used to treat NB4 and Jurkat cells for 24 h. As shown in Figure 1(A), ATO significantly enhanced the expression of PD-L1 at the protein level in NB4 and Jurkat cells compared with that in the control group cells. Due to glycosylation of the PD-L1 protein, 1-3 bands were apparent in the results. The PCR assay also indicated that the mRNA level of PD-L1 increased after ATO treatment in these cells, as shown in Figure 1(B). IFN-γ is a stimulator of PD-L1 in cancer cells. As shown in Figure 2, the expression of the PD-L1 protein in these cells was also upregulated upon stimulation with IFN-γ (100 ng/mL). Moreover, combination treatment with ATO and IFN-γ further upregulated PD-L1 expression in these cells, indicating that leukemia cells presented a stronger ability to express PD-L1 in the presence of IFN-γ. Therefore, our results suggested that ATO upregulated the expression of PD-L1 in leukemia cells at both the mRNA and protein levels.
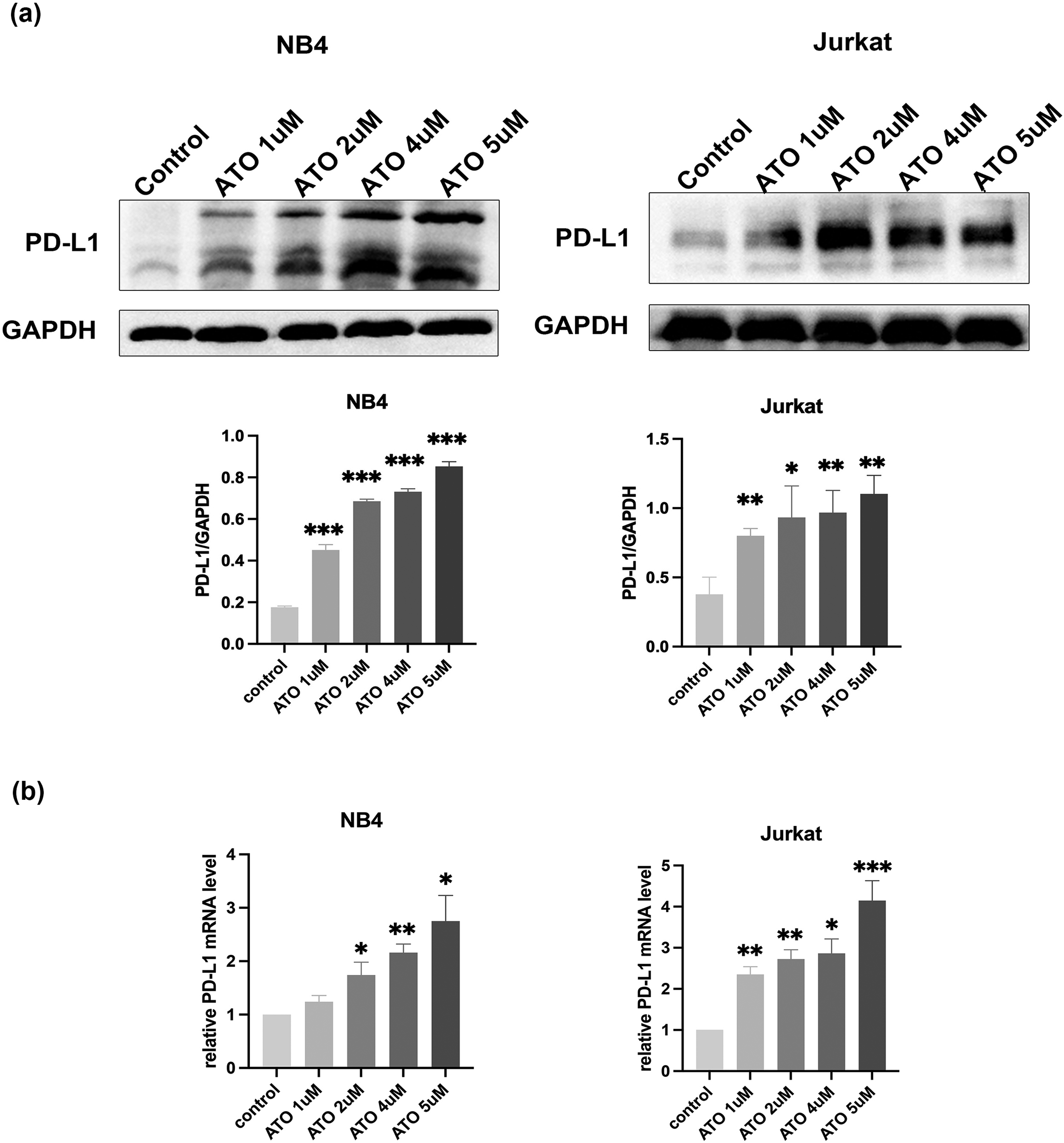
ATO-induced PD-L1 upregulation in leukemia cells. (A) and (B) The effects of ATO on the expression of PD-L1 at protein and mRNA levels in NB4 and Jurkat cells. *, ** and *** indicate P values less than .05, .01 and .001, compared with the control group. Abbreviations: ATO, arsenic trioxide; PD-L1, programed death-ligand 1.

ATO further enhanced the IFN-γ-induced expression of PD-L1 in NB4 and Jurkat cells. *, ** and *** indicate P values less than .05, .01 and .001, compared with the control group. Abbreviations: ATO, arsenic trioxide; PD-L1, programed death-ligand 1.
ATO Induced PD-L1 Expression in a NRF2-Dependent Manner
As NRF2 has been reported to favor immune evasion of solid tumor cells, we sought to determine its role in PD-L1 expression in leukemia cells. As shown in Figure 3, the nuclear NRF2 expression significantly increased in response to ATO treatment in NB4 and Jurkat cells, and this effect was positively correlated with the upregulation of PD-L1 expression. The nuclear NRF2 protein level was significantly lower after combined ATO/brusatol treatment than after ATO treatment alone. Concomitant with the inhibition of NRF2, brusatol significantly reduced ATO-induced upregulation of PD-L1 in these cells at both the protein and mRNA levels (Figure 4). Therefore, we suggest NRF2 might be involved in the mechanism by which ATO upregulates PD-L1 expression in leukemia cells.

Arsenic trioxide (ATO) increased nuclear NRF2 expression, while brusatol reduced ATO-induced nuclear NRF2 upregulation. (A) and (B) The effects of ATO and brusatol on NRF2 protein level in the nuclear fraction in NB4 and Jurkat cells. * and ** indicate P < .05 and P < .01.

Brusatol inhibited ATO-induced PD-L1 upregulation at the protein and mRNA levels. (A) and (B) The effects of brusatol on ATO-induced upregulation of PD-L1 expression at the protein and mRNA levels in NB4 and Jurkat cells. *, ** and *** indicate P values less than .05, .01 and .001, respectively. Abbreviations: ATO, arsenic trioxide; PD-L1, programed death-ligand 1.
PD-L1 is a Direct Transcriptional Target of NRF2
To determine whether NRF2 directly mediates the expression of PD-L1, we performed chromatin immunoprecipitation (ChIP). As shown in Figure 5, NRF2 bound to the promoter region of the PD-L1 gene and the binding activity significantly increased after ATO treatment compared with that of the control group. In contrast, brusatol markedly decreased this binding, and the relative binding activity was lower in the combination group than in the ATO-only treatment group. Therefore, these data suggested that ATO led to NRF2 enrichment at the PD-L1 promoter, thereby leading to the upregulation of PD-L1 expression, and that brusatol blocked this effect.

PD-L1 is a direct transcriptional target of NRF2. NB4 cells were treated with ATO, brusatol alone or their combination for 24 h, and ChIP assays were performed to detect the binding activity of NRF2 to the PD-L1 promoter. Abbreviations: ATO, arsenic trioxide; PD-L1, programed death-ligand 1; ChIP: chromatin immunoprecipitation.
Brusatol Enhanced ATO-Induced Proliferation Inhibition in Leukemia Cells
Brusatol is a sensitizer when used in combination with other traditional chemotherapies; however, there is no study on its combination with ATO in the treatment of leukemia. 17 To determine whether brusatol enhances ATO-induced cytotoxicity, NB4 and Jurkat cells were treated with each drug either alone or in combination for 24 h. As shown in Figure 6, ATO-inhibited cell proliferation in a concentration-dependent manner in these cells. The IC50 values for ATO were 2.87and 4.63 µM in the NB4 and Jurkat cells, respectively. As expected, the combination treatment significantly reduced the absolute numbers of viable cells in these cells compared with the effect of ATO treatment alone.
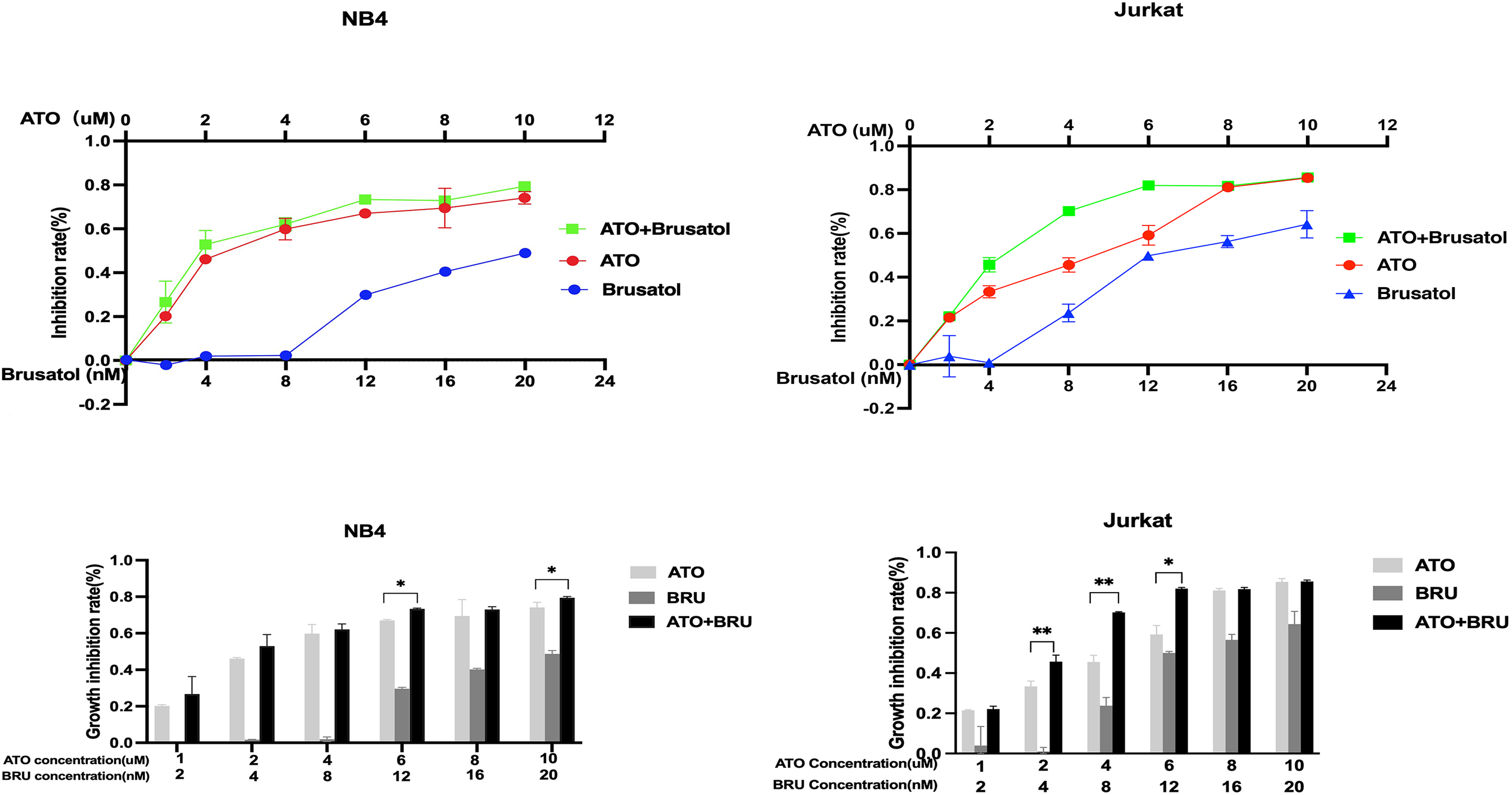
Bruastol increased arsenic trioxide (ATO)-induced proliferation inhibition in NB4 and Jurkat cells. Proliferation inhibition rates in NB4 and Jurkat cells treated with ATO, brusatol alone or their combination for 24 h. * and ** indicate P values less than .05 and .01, compared with ATO treatment alone.
Discussion
Our work indicated, for the first time, that ATO upregulated the expression of PD-L1 through NRF2 in leukemia cells, which might have weakened the antileukemia effect of ATO. Brusatol through inhibition of NRF2 expression not only decreased ATO-induced PD-L1 upregulation but also enhanced ATO-induced cytotoxicity. ATO combined with brusatol is expected to be a novel strategy in leukemia therapy.
With in-depth investigation into the pharmacological mechanism of ATO action, researchers have reported an immunosuppressive effect. Specially ATO inhibited T-cell-mediated autoimmunity by inhibiting the proliferation of T lymphocytes, inducing CD4+ T-cell apoptosis and increasing the proportion of immunoinhibitory Treg cells, and its application may improve the clinical outcome of patients with severe aplastic anemia.18,19 Additionally, Zhu et al. 9 demonstrated that ATO induced the upregulation of PD-L1 in oral squamous carcinoma cells. Consistent with previous studies, we found that ATO significantly upregulated the expression of PD-L1 at both the protein and mRNA levels in NB4 and Jurkat cells. Therefore, we suggest that ATO-induced upregulation of PD-L1 is likely to be an underlying mechanism of its immunosuppressive effect. This immunosuppressive effect of ATO may broaden its application for the treatment of autoimmune disease, but be rather harmful in cancer, where it might be partially involved in driving ATO resistance in some APL patients.
Contrary to previous findings showing that IFN-γ plays a critical role in anti-infection and tumor surveillance, recent studies have shown that lower concentrations of IFN-γ in the tumor microenvironment induced immune escape by inducing PD-L1 expression. 20 Consistent with these discoveries, our studies also found that IFN-γ upregulated PD-L1 in leukemia cells. The mechanisms involved in the regulation of PD-L1 have been extensively studied. NRF2 has been reported to be the upstream regulator of PD-L1 in melanoma. 16 Thus, we evaluated the role played by NRF2 in ATO-induced upregulation of PD-L1 in leukemia. According to our results, ATO increased nuclear NRF2 expression in NB4 and Jurkat cells and it also significantly increased the enrichment of NRF2 at the PD-L1 promoter, which could enhance the transcription of PD-L1. Brusatol reduced this enrichment via inhibition of nuclear NRF2 expression and inhibited ATO-induced upregulation of PD-L1 in leukemia cells. Taken together, the data that we report here for the first time indicate that ATO might promote immune evasion by leukemia cells via the upregulation of PD-L1 in a NRF2-dependent manner and that a combination treatment with brusatol may block this effect.
Recent studies have reported that high NRF2 expression in primary AML blasts leads to resistance to cytarabine and daunorubicin, while pharmacological inhibition of NRF2 expression by brusatol can reverse the chemoresistance.13,21 Interestingly, Banella et al. 15 noted that PML/RARα, a fusion oncogene in APL, can attenuate NRF2 transcriptional activity, which leads to better sensitivity of APL cells than cells of other subtypes of leukemia to ATO treatment. Therefore, NRF2-targeted therapy may be beneficial for durable cancer regression and prolonged survival in patients with leukemia. In the present study, the combination of ATO and brusatol led to stronger antiproliferative effects than ATO alone in leukemia cells. In particular, brusatol greatly increased the sensitivity of Jurkat cells to ATO treatment, making ATO a potential therapy for lymphocytic leukemia.
In conclusion, we propose the underlying mechanism of ATO-induced PD-L1 upregulation in leukemia cells. NRF2 inhibition by brusatol may further augment the antileukemia effects of ATO, not only by inhibiting ATO-induced PD-L1 expression but also by enhancing ATO-induced cytotoxicity. We provide a rationale for the clinical application of the combined regimen of ATO/brusatol in leukemia therapy. Further in vivo studies are warranted.
Materials and Methods
Materials and Reagents
The NB4 cell line (human APL cell line) and Jurkat cell line (human acute lymphoblastic leukemia cell line) were both purchased from BeNa Culture Collection(Henan, China). RPMI 1640 medium and fetal bovine serum (FBS) were obtained from Gibco, and ATO from Medical University Pharmaceutical Industry. Brusatol from Sigma-Aldrich was used with dimethyl sulfoxide as a solvent. Anti-GAPDH and anti-Lamin B1 were purchased from the Proteintech Group, and anti-NRF2 and anti-PD-L1 antibodies from Abcam.
Cell Culture
NB4 and Jurkat cells were cultured in RPMI 1640 medium with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37 °C.
Western Blotting
NB4 and Jurkat cells were treated with ATO, or brusatol alone or in combination. Total protein lysates were extracted using RIPA buffer on ice in the presence of a protease inhibitor cocktail (Thermo Fisher Scientific). Nuclear NRF2 was evaluated in nuclear protein lysates prepared with a nuclear and cytoplasmic protein extraction kit (Beyotime Biotechnology). Protein concentrations were assayed with a Bradford protein assay kit (Bio-Rad). Cell lysates were separated and transferred onto a 0.45 nm PVDF membrane. Then, the membranes were blocked with 5% milk-PBS-Tween followed by incubation with primary antibodies at 4 °C overnight. All primary antibodies were diluted in 5% BSA-PBS-Tween to a 1:1000 ratio. After washing with TBST 3 times, the membranes were incubated with corresponding horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, the ECL color was developed and detected with a ChemiDocTM Touch Imaging system (Bio-Rad).
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen). The RNA concentration was measured by absorbance of ultraviolet light at a wavelength of 260/280 nm using a Biophotometer. cDNA was synthesized through reverse transcription using a high-capacity cDNA reverse transcription kit (Applied Biosystems). qPCR was performed using SYBR Green PCR Master Mix (Applied Biosystems). qPCR was conducted using a SYBR solution (5 μL), primers (0.6 μL), and template DNA (4.4 μL). The reaction conditions were 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 30 s and 68 °C for 50 s The primers were purchased from Sangon Biotech. The primer sequences for the analyzed genes are shown in Table 1. β-Actin was used as an internal control.
Primer Sequences for the Analyzed Genes.

Cell Counting kit-8 Experiments
NB4 cells and Jurkat cells were seeded into 96-well plates at 2 × 105/mL and 4 × 105/mL, respectively, followed by incubation with different treatments (ATO[1-10 μM], brusatol [2-20 nM] and ATO/brusatol) for 24 h. Then 10 μL of cell counting kit 8 (CCK-8) reagent (Dojindo) was added to the cells, which were incubated for another 3 h in the dark. The absorbance was detected at an OD of 450 nm.
Chromatin Immunoprecipitation Assay
Briefly, NB4 cells were collected and cross-linked with 1% formaldehyde for 20 min at room temperature, and the reaction was stopped by adding glycine to a final concentration of 0.125 M. After washing twice in PBS, cell pellets were resuspended in ChIP buffer and incubated on ice for 30 min. Then, nuclei were pelleted at 3000 r/min at 4 °C and incubated on ice for 30 min in nuclear lysis buffer. Afterward, the lysates were sonicated to obtain DNA fragments. Immunoprecipitation was performed with an anti-NRF2 antibody (Abcam) and an anti-rabbit IgG control. The bound chromatin was pulled down by A/G agarose beads. After extensive washing, the bound DNA fragment was eluted and assessed by PCR. The primers for PD-L1 transcript variant 2, forward 5' GCACTGACATTCATCTTC 3' and reverse 5' GTTCCAATGCTGGATTAC 3' were purchased from Sangon Biotech.
Statistical Analysis
All data are represented as the mean ± standard deviation (SD). Statistical analysis for significance was performed by t-test for 2 group comparisons and 1-way ANOVA for multiple group comparisons. A P value <.05 was considered to be statistically significant. All experiments were independently repeated at least 3 times.
Footnotes
Acknowledgments
The authors would like to give their sincere gratitude to the reviewers for their constructive comments.
Data Availability
The datasets generated for the current study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.