
Abstract
Introduction
Viral pneumonia is a prevalent cause of respiratory infection that may result in the death of very young children and the elderly above the age of 75 years. 1 The emergences of avian influenza A (H5N1), severe acute respiratory syndrome (SARS), and the ongoing coronavirus infectious disease 2019 (COVID-19) have emphasized the important role of viral infection as causes of pneumonia. 2
The real-time reverse transcription polymerase chain reaction (RT-PCR) and other new rapid molecular assays for pneumonia detection are contributing to viral pneumonia diagnosis. 3 However, its treatment is undesirable on account of less active targets against viruses. Antiviral medications, such as oseltamivir, ribavirin, and acyclovir, are currently limited in clinical practice because of their side effects and virus resistance. Despite the use of immunotherapy, it has played a limited role in the treatment of viral pneumonia. 2
Traditional Chinese medicines (TCMs), such as Lianhua Qingwen (LHQW) capsule, Jinhua Qinggan (JHQG), and Lung Cleansing and Detoxifying Decoction (LCDD), have long been used for the treatment of respiratory diseases.4–6 Eupatorium lindleyanum DC. (EL), also called “Yemazhui,” has the effects of “clearing heat to remove toxicity,” and is widely used to treat cough and chronic bronchitis for various ages in China. 7 A great number of studies have proved that EL has many pharmacological activities, including anticancer,8,9 antiviral, 10 and anti-inflammatory. 11 Importantly, EL preparations, such as Eupatorium Kechuan powder and Yemazhui capsule, are clinically applied to treat respiratory diseases. 7 F1012-2 is a novel, active, sesquiterpene lactone fraction isolated from EL. In our previous study, we reported that F1012-2 had significant anticancer activities.9,12 However, its effect on viral pneumonia remains unknown.
In this study, we explored the molecular mechanism of F1012-2 against viral pneumonia using computational pharmacology methods. Network pharmacology is a novel approach that mixes computer science and medicine, establishing a “compound-protein/gene-disease” network. This method is perfect for research of TCMs that have multi-components. 13 Molecular docking methods explore the ligand conformations adopted within the binding sites of macromolecular targets, which are used in modern drug design. These techniques provide powerful tools for understanding the effect and mechanism of F1012-2 against viral pneumonia.
Materials and Methods
Obtaining the Targets of F1012-2 and Viral Pneumonia
F1012-2 consists of 3 compounds, namely Eupalinolide G (EG), Eupalinolide I (EI), and Eupalinolide J (EJ). The ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS) chromatogram of F1012-2 and the structures of its compounds can be seen in the Supplemental material. The ratio of EG, EI, and EJ in F1012-2 was 2:3:1, based on peak areas. First, the active compounds of F1012-2 and their targets were obtained from the Traditional Chinese Medicine Database and Analysis Platform (TCMSP, https://old.tcmsp-e.com/tcm-sp.php), 14 SwissTargetPrediction (https://swisstargetprediction.ch/), 15 similarity ensemble approach (SEA, http://sea.bkslab.org/), 16 and Bioinformatics Analysis Tool for the molecular mechanism of TCM (BATMAN-TCM, http://bionet.ncpsb.org.cn/batm-tcm/). 17 A compound–target network was constructed by means of Cytoscape_v3.8.2. Then, several databases were used to search for viral pneumonia-related genes: Online Mendelian Inheritance in Man (OMIM, https://omim.org/), 18 GeneCards 5.9 (https://www.genecards.org/), 19 DrugBank 5.0 (https://www.drugbank.ca/), 20 DisGeNET 7.0 (https://www.disgenet.org/), 21 and Therapeutic Target Database (TTD, http://db.idrblab.net/ttd/). 22 The target gene set was acquired with the help of Uniprot (https://www.uniprot.org/). 23 Subsequently, we took the target genes of F1012-2 and the therapeutic targets for viral pneumonia to obtain the overlapping genes.
Enrichment Analysis
Based on the overlapping genes of F1012-2 and viral pneumonia, gene ontology (GO), including biological processes (BP), cellular components (CC), and molecular functions (MF), was performed to illustrate the underlying mechanism, and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis to find the key signaling pathways.
Protein–Protein Interaction (PPI) Network and Core Subnetwork
The targets of F1012-2 and viral pneumonia were used to build a PPI network through STRING database. We set a combined score of 0.400. Afterward, the PPI network was imported into Cytoscape. We filtered genes according to each score of degree, betweenness, and closeness to acquire the critical subnetwork.
Molecular Docking Analysis
Molecular docking analysis was carried out as described previously. 24 The 2D ligand structures of EG, EI, and EJ were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov). 25 We downloaded the 3D structure of the proteins in RCSB Protein Data Bank (PDB) as .pdb files. 26 All chains of the structure were used to prepare the receptor, and all water molecules around the protein were removed. PyMOL v2.3.2 software was performed to dehydrate the receptor protein, and Autodock software was used to carry out hydrogenation and charge calculation of proteins. 27 Finally, several docking poses were obtained for the molecule by Autodock Vina, 28 and the one with the optimal binding energy was chosen.
Results
Screening the Potential Targets of F1012-2 and Viral Pneumonia
F1012-2 consists of 3 compounds, namely EG, EI, and EJ. Their structures are shown in Figure 1A. From TCMSP, SwissTargetPrediction, SEA, and BATMAN-TCM databases, we acquired 110 compound-target genes of these 3 compounds, and we visualized the compound–target interaction network by Cytoscape_v3.8.2 (Figure 1B). Then, we gained 696, 4016, 42, 13, and 5 viral pneumonia-related genes from OMIM, GeneCards, DrugBank, DisGeNET, and TTD databases, respectively (Figure 1C). After removing the duplication, 4322 genes were acquired. By overlapping the targets of the compounds and the disease, we finally obtained 78 candidate genes (Figure 1D).

Identification of compound–target gene interactions. (A) Structures of EG, EI, and EJ. (B) Construction of the 3 compounds–target interaction network. (C) Identification of the viral pneumonia-related genes by taking a union of all the results from 5 databases. (D) Identification of an intersection of compound-target genes and viral pneumonia-related genes.
GO Enrichment Analysis
To find the potential BP, CC, and MF of the 78 genes, GO enrichment analysis was carried out with Database for Annotation, Visualization and Integrated Discovery (DAVID) database. According to the Q value from small to large, the top 20 terms are shown in Figure 2. The GO analysis indicated that these genes played a vital role in various BP, such as drug response, signal transduction, apoptosis, and inflammatory response. The CC analysis indicated that plasma membrane and cytosol were the major locations where F1012-2 exerted its antiviral pneumonia activity.

Gene Ontology (GO) enrichment analysis of 78 target genes. (A) Top 20 significantly enriched biological processes (BP). (B) Top 20 significantly enriched cellular components (CC). (C) Top 20 significantly enriched molecular functions (MF).
KEGG Enrichment Analysis
To understand further how F1012-2 affects viral pneumonia through these target genes, KEGG enrichment analysis was performed by importing the 78 target genes into the DAVID database. We selected the top 20 terms based on the Q value from small to large, as shown in Figure 3. The analysis indicated that these target genes mainly affected the pathways related to cancer, sphingolipid, neurotrophin, tumor necrosis factor (TNF), and Toll-like signal pathways. In addition, we found other virus-related signaling pathways, as shown in Table 1, suggesting that these play an important role in virus infection.

Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the top 20 significantly enriched pathways.
Virus-Related Pathway Enriched by 78 Target Genes With Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis.

PPI Network and key Network Analysis
A PPI network of 78 targets was first obtained from STRING database, and then imported into Cytoscape for further analysis. The filters were set as an adjusted average of degree, betweenness, and closeness. Finally, 5 key target genes epidermal growth factor receptor (EGFR), interleukin-1beta (IL1B), tyrosine-protein kinase SRC, Caspase-3 (CASP3), and transcription factor and oncoprotein JUN were acquired (Figure 4).

Identification of key target genes by Cytoscape.
Molecular Docking of Active Compounds and the key Proteins
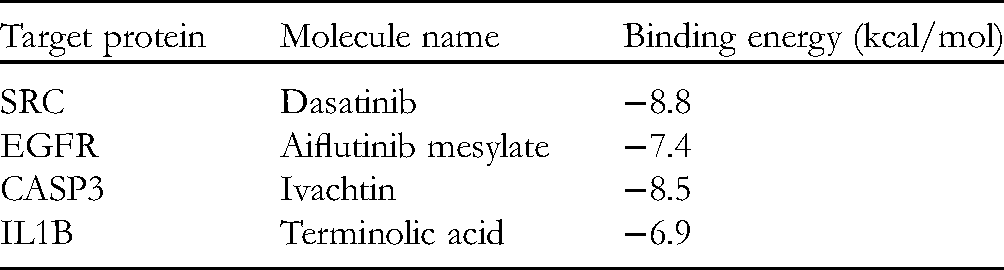
Five key target genes, EGFR, IL1B, SRC, CASP3, and JUN, were selected to dock with EG, EI, and EJ. The docking scores were shown in Figure 5A. We set the docking score at −6.0 kcal/mol. The blue color, with a high docking score, indicates a poor connection needing high binding energy. Conversely, the red color, with a low docking score, indicates a better interaction needing low binding energy. As shown in Figure 5A, the docking scores of JUN with EG, EI, and EJ were all higher than −6.0 kcal/mol indicating that EG, EI, and EJ presented lower binding activities with JUN. The docking scores of EGFR, IL1B, SRC, and CASP3 with EG, EI, and EJ were all lower than −6.0 kcal/mol indicating that the 3 compounds could easily bind in the pockets of EGFR, IL1B, SRC, and CASP3 proteins. Therefore, we exhibited the molecular docking of EGFR, IL1B, SRC, and CASP3 with EG, EI, and EJ in Figure 5B. In order to investigate further the molecular docking of the active compounds with EGFR, IL1B, SRC, and CASP3, the inhibitors of these 4 key proteins (namely Dasatinib, Alflutinib mesylate, Ivachtin, and terminolic acid) were used as positive controls. The results showed that the binding energies of the positive controls with the target proteins were all lower than −6.0 kcal/mol (Table 2).

Molecular docking of active compounds and key proteins. (A) Molecular docking score. (B) Molecular docking between Eupalinolide G (EG), Eupalinolide I (EI), and Eupalinolide J (EJ) ligands and SRC, CASP3, EGFR, and IL1B receptors.
The Binding Energy of the Target Protein With Their Positive Controls by Molecular Docking Analysis.
Discussion
Viral pneumonia is extremely infectious and pathogenic. For example, COVID-19 has rapidly spread around the world over the past 2 years, and there is still a lack of targeted drugs. TCM has a long history of treating epidemic disease, known as “Wen Yi” or “Wen Bing” in ancient China. TCMs consist of various herbs or natural products with many active compounds that target multiple pathways and are applied for the different symptoms of a patient. 29 However, it is difficult to elucidate the exact mechanism of a TCM due to its diverse compounds and their targets.
Network pharmacology is a new subject that is based on the theory of computer science, medicine, and systems biology, which has been widely used for TCM. 30 As an important and assistant method, network pharmacology can better understand the interaction mechanism of the multicomponents and multi-targets of TCM. EL is traditionally used to treat respiratory disease, and made a great contribution in the fight against SARS-CoV in 2003. 31 In this present study, we conducted an EL target and viral pneumonia-related gene intersection that acquired a total of 78 target genes by analyzing the active fraction F1012-2 of EL. GO and KEGG enrichment analysis indicated that EL can regulate pathways related to virus pneumonia. Among the 78 target genes, PPI network and network analyses found 5 key targets. Molecule docking was performed to verify further the interaction between the compounds of EL and the key targets.
The enrichment analysis of the target genes of EL's interaction with viral pneumonia by GO and KEGG is interesting. First, the 8 significant KEGG terms indicated that EL could regulate viral infection. This result was consistent with a previous study 10 which indicated that EL had antiviral effects. Moreover, the GO (BP) terms indicated that EL might be involved in the process of apoptosis and anti-inflammation during the treatment of viral pneumonia.
The active fraction F1012-2 is composed of 3 distinct compounds, EG, EI, and EJ, which have been reported to possess anti-inflammatory, 32 and antitumor33,34 activities. However, the antiviral pneumonia effect has not been investigated. Molecular docking can provide further information of the interaction between the compounds of EL and the key targets. In our study, we found that EG, EI, and EJ could easily bind into the pocket of EGFR, IL1B, SRC, and CASP3, and EJ had the best binding activity with lower binding energy (−8.3, −7.4, −9.1, and −7.4 kcal/mol) than EG (−7.0, −7.1, −7.8, and −7.5 kcal/mol) and EI (−7.2, −7.5, −7.4, and −7.0 kcal/mol), as shown in Figure 5A. In order to investigate the reliability of molecular docking, we examined the molecular docking of the four target proteins with their positive controls. The binding energies of the positive controls with the target proteins were all lower than −6.0 kcal/mol (Table 2). Furthermore, the binding energy of EJ with SRC (−9.1 kcal/mol), EGFR (−8.3 kcal/mol), and IL1B (−7.4 kcal/mol) were all lower than the positive control (−8.8, −7.4, and −6.9 kcal/mol, respectively), as shown in Figure 5A and Table 2, thus suggesting that EJ has a better connection with SRC, EGFR, and IL1B. Therefore, based on the above results, we proposed that EJ might be the main active component of F1012-2. In addition, among the key targets of SRC, EGFR, and IL1B, EJ and SRC have the best connectivity. The proteases play a key role in viral replication. SRC, a known regulator of cellular tyrosine kinase, has an important role in cell mobility, proliferation, and survival, 35 and is the main target in cellular cleavage events. Some SRC inhibitors are in current clinical use. 36 These results showed that EJ might be one of the SRC inhibitors.
In this network-based study, we explored the potential therapeutic targets and signal pathways of EL against viral pneumonia. The major pathways involved were sphingolipid, neurotrophin, TNF, and Toll-like receptor signal pathways. EGFR, IL1B, SRC, and CASP3 were the key targets. These multi-target and multi-pathways can be applied to illustrate the potential mechanisms of EL's effect on viral pneumonia, which is constructive to the development of new therapeutic strategies against viral pneumonia using the Chinese herb EL. Nevertheless, further experimental studies are needed to validate this assumption.
Conclusion
We have suggested the potential mechanism of EL against viral pneumonia using computational pharmacology. GO and KEGG enrichment analysis showed that the major pathways involved were sphingolipid, neurotrophin, TNF, and Toll-like receptor signaling pathways. Molecular docking verified EGFR, IL1B, SRC, and CASP3 as the key targets, and EJ might be the main active component of F1012-2. We believe that this discovery may contribute to the development of new therapeutic strategies against viral pneumonia.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221131410 - Supplemental material for Prediction Targets of a Novel Sesquiterpene Lactone From Eupatorium lindleyanum DC. Against Viral Pneumonia Using Computational Pharmacology
Supplemental material, sj-docx-1-npx-10.1177_1934578X221131410 for Prediction Targets of a Novel Sesquiterpene Lactone From Eupatorium lindleyanum DC. Against Viral Pneumonia Using Computational Pharmacology by Xintong Shen, Hongtao Hu, Jiajin Xu and Shasha Tian in Natural Product Communications
Footnotes
Acknowledgements
The authors are extremely thankful to Dr Siyue Lou from Zhejiang Chinese Medical University for kindly revising the language of the article.
Author’s Contribution
Xintong Shen and Hongtao Hu performed the statistical analysis. Shasha Tian and Jiajin Xu drafted the article and Xintong Shen and Hongtao Hu were co-first authors. All authors contributed to the article and approved the submitted version.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data are accessible once requested from the corresponding author.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Key Research Project of Traditional Chinese Medicine in Zhejiang Province, Zhejiang Chinese Medical University Research Fund Project, College Students’ innovation project, and Natural Science Foundation of Zhejiang Province (grant numbers 2022ZZ008, KC201912, S202210344108, and LQ19H160006).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.