
Abstract
Keywords
Introduction
Coronary artery disease (CAD) is a lethiferous disease that accounts for 13.2% of all deaths worldwide. 1 CAD is a major cause of myocardial ischemia or myocardial infarction (MI). Currently, the most effective therapeutic interventions to alleviate acute myocardial ischemic injury or to control the size of MI are primary percutaneous coronary interventional intervention or thrombolytic therapy, with timely and effective reperfusion. 2 But the process of reperfusion can lead to adverse effects on cardiomyocytes, which is known as myocardial ischemia-reperfusion (I/R) injury. 3 Previous studies have provided evidence for the improvement of clinical outcomes in patients with acute MI by preventing myocardial I/R injury. 2
Ferroptosis, a recently recognized cell death distinct from apoptosis, has been found to be associated with many disease states. 4 Ferroptosis is caused by the accumulation of lethal levels of toxic lipid hydroperoxides (iron dependent), which may attribute to an imbalance between oxidative mechanism (hyperactivity) and antioxidant mechanism (weak). 5 Recent investigations have demonstrated the involvement of ferroptosis in I/R injury in several organs, given that both I/R injury and ferroptosis are closely related to cellular oxidative damage. 6 Inhibition of ferroptosis has been documented to be beneficial for I/R conditions, which implies that ferroptosis may be a potential target for the MI therapeutic strategies.6-8
Pratensein (PTS) is an isoflavone compound isolated from Trifolium pretense L. with considerable anti-inflammatory activity and antioxidant activity. In previous studies, PTS ameliorates β-amyloid-induced oxidative damage by inhibiting the activation of NF-κB, which strongly caused cognitive impairment in rats. 9 Chen et al. 10 evaluated the protective effect of five isoflavones (calycosin, daidzein, formononetin, irilone, and PTS) from T pretense on neurons against LPS-induced production of inflammatory and oxidative mediators, and they found that PTS exerted the highest protective potency. In addition, flavonoids are known to be effective cardioprotective drugs. For example, Quercetin (a common flavonoid) and its derivatives have potential beneficial effects in different cell cultures and animal models of cardiac injury. 11 Furthermore, Dracocephalum moldavica L. (also known as Xinjiang, China) is a herb rich in flavonoids, 12 that has been shown to alleviate apoptosis induced by I/R injury. 13 Similarly, an isoflavone from T pretense, biochanin A (BCA) attenuates myocardial I/R injury by blocking the inflammatory response in rats. 14 However, the protective effect of PTS on myocardial I/R injury was unclear.
In a number of experimental studies, the oxygen-glucose deprivation/reperfusion (OGD/R) model was used to mimic the myocardial I/R injury.15-17 Here, we also used the OGD/R model in H9c2 cells to explore the function of PTS and the related mechanism.
Materials and Methods
H9c2 Cells Culture
Rat embryonic ventricular myocardial cell line H9c2 cells were cultured in DMEM (ThermoFisher) containing 5% CO2 in humidified air. To construct the OGD/R model, H9c2 cells were cultured in serum-free and glucose-free media, exposed to hypoxia conditions (1% O2, 5% CO2, and 94% N2) for 4 h, and then reoxygenated under hyperoxia condition (21% O2, 5% CO2, and 74% N2) for 24 h.
SiRNAs Transfection
For transfection of siRNAs, cells were transfected with either si-Nrf2 or si-NC (Ribobio) prior to OGD/R treatment with Lipofectamine 3000 reagent (ThermoFisher).
Cell Counting Kit-8 Assay
H9c2 cell viability assay was conducted after plating in a 96-well plate at a density of 5000 (cells per well). The Cell Counting Kit-8 (CCK-8) solution (10 μL; Sigma) was added to cells for incubation of 4 h. The absorbance at 450 nm was measured using a microplate reader (Molecular Devices).
The TdT-Mediated dUTP-Biotin Nick end Labeling
For TdT-mediated dUTP-biotin nick end labeling (TUNEL) assay, cells were fixed in 4% paraformaldehyde for 30 min and then incubated with 0.3% Trition X-100 for 5 min. Cells were then incubated with TUNEL detection buffer (Beyotime) for 1 h. DAPI was used for nucleus staining. The cells were observed under a fluorescence microscope (Leica DM1000) and the TUNEL-positive cells were counted.
Flow Cytometry Assay
Cell apoptosis was determined using the Annexin VFITC Apoptosis Detection Kit (Vazyme) as directed by the manufacturer. In brief, cells were washed with cold PBS and resuspended in binding buffer and fixed, then incubated for 10 min in the dark with 5 μL Annexin V-FITC and 5 μL 7-AAD staining solution at room temperature. After mixing with 400 μL 1 × binding buffer, apoptosis was examined with FACS Calibur flow cytometer (Bection Dickinson) within 1 h. 18
Reactive Oxygen Species Detection
To detect reactive oxygen species (ROS) production, cells were incubated with probe DCFH-DA (10 μM; Beyotime) for 25 min. Cells were then rinsed twice with PBS and imaged with a fluorescence microscope. The fluorescence intensity was examined using a fluorescence microplate reader (excitation/emission 488/525 nm).
Measurement of Malondialdehyde, Glutathione, and Fe2+
The cells lysates were prepared to determine the malondialdehyde (MDA), glutathione (GSH), and Fe2+ levels using the corresponding Assay Kits (Jiancheng Bio.). Briefly, the MDA level was measured using the TBA method, and the absorbance of red products was read at the wavelength of 530 nm. The reduced GSH level was assessed using DTNB method. The yellow products formed by DTNB and GSH were measured by microplate reader at the wavelength of 405 nm. The Fe2+ level was tested by detecting the absorbance of its complexes with 2,2'-bipyridine at 520 nm.
Western Blotting Analysis
Total and nuclear proteins were extracted with a commercial kit (Beyotime), followed by a BCA protein assay (Solarbio). After that, 40 μg of total protein was subjected to 12% SDS-PAGE electrophoresis and western blotting analysis using the conventional method. 19 Primary antibodies against Nrf2, GPX4, Lamin B1, and α-tubulin, as well as HRP-linked IgG secondary antibodies, were acquired from Santa Cruz Bio (Santa Cruz). Image Lab Software (Bio-Rad) was used to analyze the intensity of protein bands.
Data Analysis
All values were analyzed using GraphPad Prism 8 statistical software and expressed as the mean ± SD. Comparisons of data among multiple groups were performed using one-way ANOVA and post hoc Tukey's multiple comparison test. P < .05 was considered statistically significant.
Results
Effect of PTS on H9c2 Cells Viability
H9c2 cells were exposed to PTS (structure in Figure 1A) (0, 10, 20, 50 μM) for 48 h. Cell viability decreases significantly with incubation of PTS (20 and 50 μM), while in H9c2 cells treated with PTS (10 and 20 μM), cell viability remains virtually unchanged (Figure 1B). For the following tests, concetrations of 10 and 20 μM were used.

Changes in cell viability of H9c2 cells with PTS exposure (0, 10, 20, 50 μm). (A) Chemical structure of PTS. (B) H9c2 cells were subjected to PTS exposure (0, 10, 20, 50 μM) for 48 h, followed by the determination of cell viability using CCK-8 assay. “***” indicates significantly statistical differences versus control cells with P < .001. Abbreviations: CCK-8, Cell Counting Kit-8; PTS, pratensein.
PTS Alleviates OGD/R Induced Apoptosis of H9c2 Cells
TUNEL-positive cells were selected as representative of apoptotic cells (Figure 2A). The TUNEL-positive cells were counted to calculate the apoptotic cells. As shown in Figure 2B, OGD/R-treated H9c2 cells had significantly higher apoptotic cells than control cells, which can be counteracted by PTS (10 and 20 μM). As shown in Figure 2B and 2C, OGD/R-induced apoptosis of H9c2 cells was exacerbated compared to the control cells, and PTS (10 and 20 μM) can reverse it. At the same time, the cell viability of OGD/R-treated H9c2 cells was markedly decreased. However, PTS treatment (10 and 20 μM) improved the damaged cell viability of OGD/R-treated H9c2 cells (Figure 2D).

Changes in cell apoptosis of H9c2 cells after indicated treatments. (A), (B) Cell apoptosis of H9c2 cells was assessed by TUNEL assay, and TUNEL-positive cells were selected as representative of apoptotic cells. Representative images of TUNEL assay were presented and the TUNEL-positive cells were counted to calculate the percentage of apoptotic cells (A). (B), (C) Cell apoptosis of H9c2 cells was examined by flow cytometer analysis. (D) Cell viability of H9c2 cells was determined using CCK-8 assay. “***” indicates significantly statistical differences versus control cells with P < .001. “&&” and “&&&” indicate significantly statistical differences versus OGD/R-treated cells with P < .01 and P < .001, respectively. Abbreviations: TUNEL, TdT-mediated dUTP-biotin nick end labeling; CCK-8, Cell Counting Kit-8; OGD/R, oxygen-glucose deprivation/reoxygenation.
PTS Alleviates OGD/R Induced Oxidative Stress and Inhibits Ferroptosis in OGD/R-Insulted H9c2 Cells
Figure 3A and B showed that ROS production in OGD/R-treated H9c2 cells was 2.8-fold higher than that in normal cultured H9c2 cells. While the increase in ROS production was attenuated by PTS (10 and 20 μM). In addition, OGD/R treatment led to a significant increase in MDA level and a decrease in GSH level, which can be mitigated by PTS (10 and 20 μM; Figure 3C and D). In OGD/R-treated H9c2 cells, Fe2+ level increased obviously by 3.8-fold, while PTS (10 and 20 μM) treatment prevented the OGD/R-induced increase in Fe2+ level (Figure 3E).
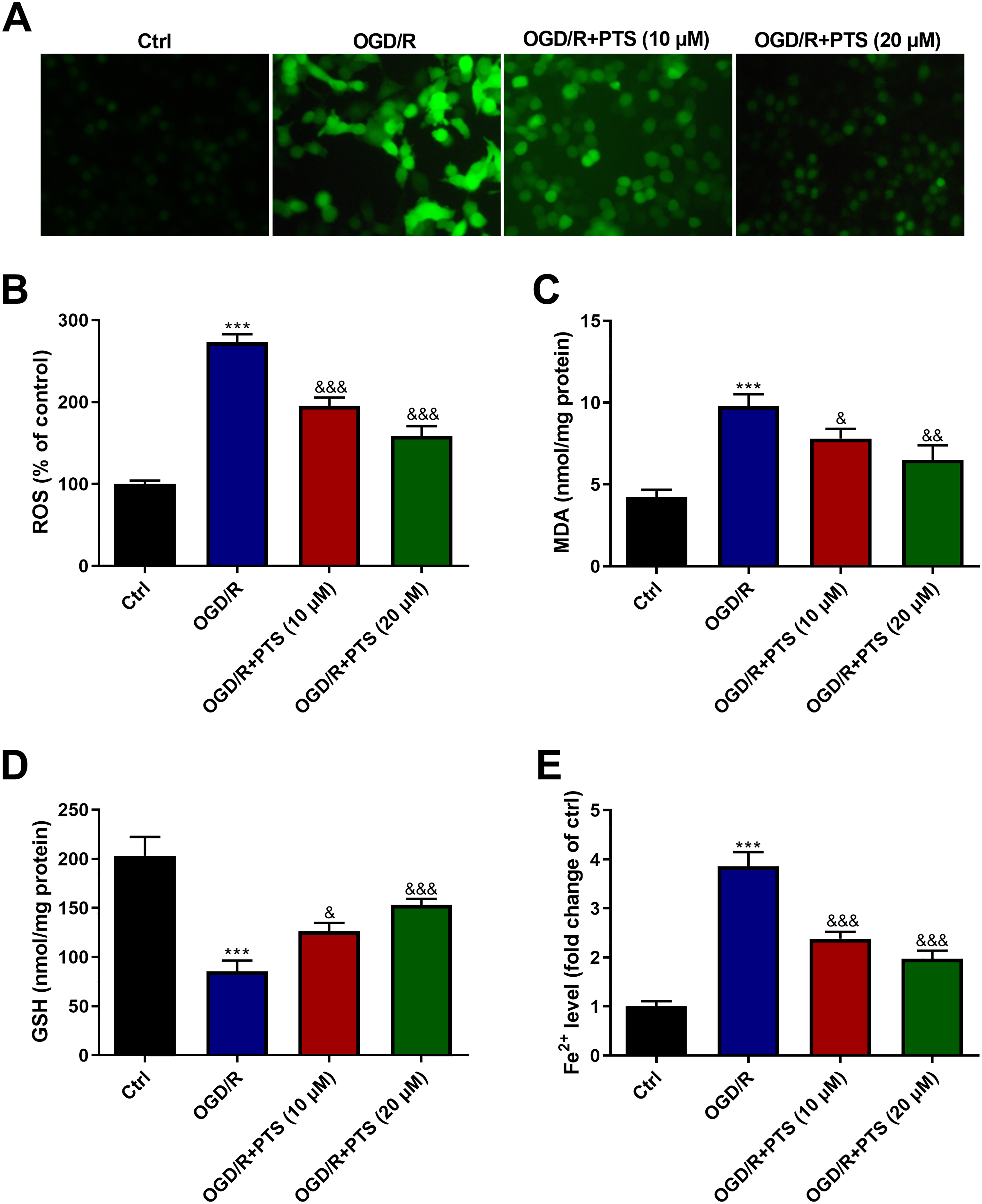
Changes in oxidative stress and ferroptosis of H9c2 cells after indicated treatments. (A, (B) ROS production in H9c2 cells was determined using a probe DCFH-DA. Representative images were captured using a fluorescence microscope (A), and the fluorescence intensity was examined using a fluorescence microplate reader (B). (C), (D) MDA level and GSH content in H9c2 cells were determined to reflect oxidative status. (E) Fe2+ level in H9c2 cells was examined to indicate ferroptosis. “***” indicates significantly statistical differences versus control cells with P < .001. “&”, “&&”, and “&&&” indicate significantly statistical differences versus OGD/R-treated cells with P < .05, P < .01, and P < .001, respectively. Abbreviations: GSH, glutathione; OGD/R, oxygen-glucose deprivation/reoxygenation; ROS, reactive oxygen species; MDA, malondialdehyde.
PTS Activates the Nrf2/GPX4 Axis in OGD/R-Insulted H9c2 Cells
Next, we used the western blotting technique to assess the participation of the Nrf2/GPX4 signaling in H9c2 cells. The data shown in Figure 4A indicated that Nrf2 expression was markedly reduced by 62.7% in OGD/R-treated H9c2 cells. PTS (10 and 20 μM) treatment upregulated Nrf2 expression with 1.59- and 2.04-fold changes. Figure 4B suggested that the OGD/R treatment obviously downregulated the expression of GPX4 in H9c2 cells, which can be restrained by PTS (10 and 20 μM).

Changes in the Nrf2/GPX4 signaling pathway in H9c2 cells after indicated treatments. Western blotting technique to evaluate the involvement of the Nrf2/GPX4 signaling in H9c2 cells by detecting the expression levels of Nrf2 (A) and GPX4 (B). “***” indicates significantly statistical differences versus control cells with P < 0.001. “&”, “&&”, and “&&&” indicate significantly statistical differences versus oxygen-glucose deprivation/reoxygenation (OGD/R)-treated cells with P < .05, P < .01, and P < .001, respectively.
Downregulation of Nrf2 Attenuates the Decreased Cell Apoptosis in PTX-Treated H9c2 Cardiomyocytes
We next explored the effect of Nrf2 downregulation on H9c2 cells with various treatments by transfecting si-Nrf2. Figure 5A and B indicated a markedly increase in TUNEL-positive cells in H9c2 cells with downregulated Nrf2 compared to H9c2 cells treated with PTS (20 μM). Similarly, apoptosis of H9c2 cells with downregulated Nrf2 increased significantly compared to H9c2 cells treated with 20 μM PTS (Figure 5B and C). Results in Figure 5D implied that the improvement of cell viability caused by PTS (20 μM) was restrained by si-Nrf2 transfection.

Changes in the protective effect of PTX against cell apoptosis in OGD/R-insulted H9c2 cells after downregulation of Nrf2. Nrf2 was knocked down in H9c2 cells through transfection with si-Nrf2. (A), (B) Cell apoptosis of H9c2 cells was assessed by TUNEL assay, and TUNEL-positive cells were selected as representative of apoptotic cells. Representative images of TUNEL assay were presented and the TUNEL-positive cells were counted to calculate the percentage of apoptotic cells (A). (B) (C) Cell apoptosis of H9c2 cells was examined by flow cytometer analysis. (D) Cell viability of H9c2 cells was determined using CCK-8 assay. “***” indicates significantly statistical differences versus control cells with P < .001. “&&&” indicates significantly statistical differences versus OGD/R-treated cells with P < .001. “##” and “###” indicate significantly statistical differences versus (OGD/R + PTS + si-NC)-treated cells with P < .01 and P < .001, respectively. Abbreviations: OGD/R, oxygen-glucose deprivation/reoxygenation; TUNEL, TdT-mediated dUTP-biotin nick end labeling; CCK-8, Cell Counting Kit-8; PTS, pratensein.
Downregulation of Nrf2 Attenuates the Decreased Oxidative Stress and Ferroptosis in PTX-Treated H9c2 Cardiomyocytes
The data in Figures 6A to C showed that the inhibition of ROS and MDA by PTS (20 μM) was blocked by si-Nrf2 transfection. As illustrated in Figure 6D, the increase in GSH content due to PTS (20 μM) was also restrained by the downregulation of Nrf2. In addition, reduced Fe2+ levels in H9c2 cells treated with PTS (20 μM) were markedly elevated after transfection with si-Nrf2 (Figure 6E).

Changes in the protective effect of PTX against oxidative stress and ferroptosis in OGD/r-insulted H9c2 cells after downregulation of Nrf2. (A), (B) ROS production in H9c2 cells was determined using a probe DCFH-DA. Representative images were captured using a fluorescence microscope (A), and the fluorescence intensity was examined using a fluorescence microplate reader (B). (C), (D) MDA level and GSH content in H9c2 cells were determined to reflect oxidative status. (E) Fe2+ level in H9c2 cells was examined to indicate ferroptosis. “***” indicates significantly statistical differences versus control cells with P < .001. “&&&” indicates significantly statistical differences versus OGD/R-treated cells with P < .001. “#”, “##” and “###” indicate significantly statistical differences versus (OGD/R + PTS + si-NC)-treated cells with P < .05, P < .01 and P < .001, respectively. Abbreviations: GSH, glutathione; OGD/R, oxygen-glucose deprivation/reoxygenation; ROS, reactive oxygen species; PTS, pratensein.
Discussion
We firstly found that PTS protected H9c2 cells from OGD/R-induced apoptosis, indicated by reduction of TUNEL-positive cells and elevation of cell viability after PTS treatment. Then, we attempted to explore its potential mechanisms. I/R injury is usually attributed to nitrosation and oxidative stress, which are caused by increased ROS production and mitochondria dysfunction.20,21 Another type of damage is the imbalance of iron homeostasis, showing a dramatical increase in free iron level within cardiomyocytes. Free iron is predominantly utilized in mitochondria to synthesize the heme and iron-sulfur clusters. 22 Therefore, mitochondria dysfunction under I/R condition exacerbates iron accumulation. In turn, metal ion also promotes the production of ROS through the Fenton reaction, which ultimately leads to cell damage and induces activation of multiple cell death pathways, including ferroptosis. 23 In fact, the morphological feature of ferroptosis is morphologically characterized by changed mitochondria structure. 24 In addition, since cardiomyocytes consume plentiful intracellular ATP (more than 90%), mitochondrial function is quite crucial for the heart tissue to obtain energy. 25 For this reason, ferroptosis is quite an essential mechanism for I/R-induced cardiomyocyte death. Based on these concepts, we speculated that PTS may protect H9c2 cells from OGD/R-caused injury by preventing ferroptosis.
Ferroptosis is iron-dependent and distinctly different from other forms of necrosis, such as necroptosis and pyroptosis. 26 Mechanisms governing ferroptosis include deregulation of iron homeostasis, ROS accumulation, cysteine and GSH metabolism, and failure of phospholipid peroxidase GPX4 and lipid peroxidation.27,28 Iron is an essential component of metalloproteins such as hemoglobin, myoglobin, and ribonucleotide reductase in almost all living cells. As a result, it is involved in multiple critical biochemical processes, including oxygen transport, cellular respiration in mitochondria, oxygenation of muscles, enzymatic reactions, as well as synthesis and repair of DNA. 29 However, excessive iron can disrupt redox homeostasis and lead to the formation of ROS, which is a potentially harmful intermediates. 30 It has been widely appreciated that ROS act as a second messenger in cell signaling. ROS disrupts the antioxidant system and triggers oxidative stress. ROS interacts with polyunsaturated fatty acids and induces lipid peroxidation. 27 These events lead to GSH depletion, which can lead to inactivation of GPX4 and eventually ferroptosis. Here, we demonstrated that the OGD/R-caused increase in the production of ROS and MDA (a product of lipid peroxidation), decrease in GSH, and increase in Fe2+ level were attenuated by PTS. Based on these findings, we speculated that PTS may protect H9c2 cells from OGD/R-caused ferroptosis.
Nrf2 is an antioxidant transcription factor that protects cells from oxidative damage by modulating the antioxidant response pathway. 31 Nrf2 occupies a significant position in the myocardial I/R injury and acts as a target for MI prevention.32,33 Notably, Nrf2 was found to directly or indirectly regulate intracellular free iron content, mitochondrial function, GPX4 protein content, and NAPDH regeneration, thereby regulating ferroptosis. 34 There is sufficient evidence to support that the Nrf2/GPX4 signaling pathway can protect cells from I/R injury via ferroptosis mechanism. Xu et al. 35 reported that naringenin alleviates myocardial I/R injury by modulating the Nrf2/GPX4 axis to inhibit ferroptosis. Kaempferol ameliorates OGD/R-induced neuronal ferroptosis by inducing activation of the Nrf2/SLC7A11/GPX4 Axis. 36 Nrf2 prevents acute lung injury by regulating GPX4, SLC7A11, and HO-1 to inhibit ferroptosis caused by intestinal I/R. 37 Here, we reported that PTS activated the Nrf2/GPX4 signaling in H9c2 cardiomyocytes under the OGD/R condition. Knockdown of Nrf2 in H9c2 cells restrained the protective effect of PTS on OGD/R-induced ferroptosis in H9c2 cardiomyocytes, implying that Nrf2 signaling mediated the cardioprotective effect of PTS.
Conclusions
In conclusion, this study provided evidence for the protective effect of PTS on OGD/R-induced ferroptosis in cardiomyocytes. In addition, the biological function of PTS was attributed to the activation of the Nrf2/GPX4 signaling. These findings may be meaningful for developing a potential therapeutic strategy for MI.
Footnotes
Authors’ Contributions
Bin Wang made a majority contribution to the conception of this study and prepared the first draft of this manuscript. Bin Wang, Wei Ma, and Yali Di carried out experiments. Yali Di performed the statistical analysis. All authors have read and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Date Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.