
Abstract
Introduction
Osteoarthritis (OA) is a progressive chronic disorder primarily affecting the knee, characterized by the gradual degeneration of joint cartilage, ossification of subchondral bone, and the formation of osteophytes. 1 In clinical practice, the main symptoms observed are the progressive development of joint pain, swelling, and stiffness. Currently, it is estimated that over 240 million people worldwide have symptomatic OA, with approximately 35% of individuals over 60 years old being affected.2,3 The pathogenesis of OA is highly complex, and existing therapeutic interventions for OA primarily focus on alleviating symptoms rather than providing safe and effective medications for delaying or reversing the progression of the disease. 4
Ferroptosis is a unique form of cell death characterized by iron-dependent mechanisms, inactivation of glutathione peroxidase 4 (GPX4), and accumulation of lipid peroxides.5,6 Unlike apoptosis, autophagy, necroptosis, and other forms of cell death, ferroptosis plays a role in the pathogenesis of various diseases, including tumors, 7 ischemic organ injury, 8 neurodegenerative disorders, 9 hematological diseases, 10 and kidney injury. 11 Recent clinical research has highlighted the significant impact of ferroptosis on the progression of OA. 12 Decreased protein levels of GPX4, which is substantially lower in OA-affected cartilage compared to healthy cartilage, has been associated with worsened OA progression. 13 Moreover, nuclear factor-erythroid 2-related factor 2 (Nrf2), which positively regulates GPX4 protein levels, 14 has shown chondroprotective effects in OA therapies. 15 Therefore, targeting the Nrf2-mediated ferroptosis pathway holds potential as a therapeutic approach for treating OA.
Quercetin (QUE), an extensively researched flavonoid compound, 16 is primarily found in various vegetables, fruits, and Chinese herbs. It possesses various biochemical and pharmacological effects, including anti-inflammatory, 17 antioxidant, 18 anti-allergic, 19 antiviral, 20 anti-cancer, 21 and anti-tumor 22 properties. Notably, QUE has demonstrated therapeutic effects in conditions such as acute kidney injury, 23 type 2 diabetes, 24 and epilepsy 25 through its regulation of ferroptosis. Furthermore, QUE has been reported to exhibit antioxidant activities by modulating the Nrf2/ heme oxygenase-1 (HO-1) pathway in the context of OA. 26 However, the potential regulatory role of QUE in ferroptosis in chondrocytes has not been explored. Therefore, we hypothesize that QUE may suppress ferroptosis in chondrocytes by modulating the Nrf2/HO-1 pathway.
This research has revealed the potential relationship between Nrf2 signaling and ferroptosis in chondrocytes, as well as the anti-ferroptosis effect of QUE in RAS-selective lethal 3 (RSL3)-induced C28/I2 cells. These findings provide valuable insights for future drug design and development strategies, opening up new possibilities in this field.
Materials and Methods
Chemicals and Reagents
QUE (S25567) was provided by Yuanye Biotechnology (Shanghai, China). RSL3 (HY-100218A), Fer-1 (HY-100579), TBHQ (HY-100489), and ML385 (HY-100523) were obtained from MCE (Shanghai, China). The primary antibody for GPX4 (T56959) was provided by Abmart (Shanghai, China), the primary antibodies for Nrf2 (80593-1-RR), HO-1 (10701-1-AP), β-actin (66009-1-Ig), and Lamin B1 (12987-1-AP) by Proteintech (Wuhan, China), the secondary antibodies, both HRP-conjugated anti-rabbit IgG (ZB-2301) and anti-mouse IgG (ZB-2305) by Zhongshan Bioengineering (Beijing, China), and the CoraLite594-conjugated Goat Anti-Rabbit IgG (SA00013-4) secondary antibody by Proteintech (Wuhan, China).
Cell Culture
The C28/I2 human chondrocyte line (STR Authentication no. 20210312-3) was provided by Chengong Biotechnology (Shanghai, China). The C28/I2 cells were cultivated in a humidified incubator at 37 °C with 5% CO2 using media consisting of DMEM (Gibco, USA), 10% FBS (Gibco, USA), and 1% antibiotic-antimycotic solution (NCM, China).
Cell Viability Assay
Chondrocytes were seeded in 96-well plates at a density of 2 × 103 cells per well. The cells were incubated with RSL3, QUE, Fer-1 (10 µM),27,28 TBHQ (10 µM),29,30 or ML385 (10 µM).31,32 After 12, 24, or 48 h, a working solution of CCK-8 (APExBIO, USA) was added to each well. Absorbance at 450 nm was measured using a microplate reader (TECAN, Switzerland).
Lipid Reactive Oxygen Species (ROS) Assay
Cells were seeded at a density of 2 × 104 cells/well in 12-well plates and 1 × 105 cells/dish in 60-mm dishes. The cells were treated with QUE (1 µM), Fer-1 (10 µM), TBHQ (10 µM), or ML385 (10 µM) individually or in combination with RSL3 (0.5 µM). Subsequently, the cells were incubated with 5 µM C11-BODIPY 581/591 (Thermo Fisher Scientific, USA). In 12-well plates, the cells were washed with HBSS (Solarbio, China), and images were captured using a fluorescence microscope (Leica Thunder, Germany) with FITC and RHOD filters (Ex 488 nm and 565 nm, Em 505-550 nm and >580 nm). Cells seeded in 60-mm dishes were harvested by trypsinization and analyzed using a Gallios Flow Cytometer (Beckman, USA) with a FITC filter (Ex 488 nm, Em 505-550 nm). A primary gate based on a dot plot of forward scatter (FSC) versus side scatter (SSC) was set to exclude dead cells or small debris, and the data were analyzed using FlowJo Software.
Malondialdehyde (MDA) Assay
Cells seeded in 60-mm dishes were treated following the same procedure as described above. Protein lysates were collected using radio immunoprecipitation assay (RIPA) lysis buffer (NCM, China). To measure MDA, 100 μL of the sample or standard solution was mixed with 200 μL of MDA detection working buffer (Beyotime, China). The mixture was then heated in boiled water and subsequently cooled and centrifuged. The absorbance was measured at 532 nm. The protein concentration determined by the Bicinchoninic Acid (BCA) Assay (Beyotime, China) was used to normalize the MDA content.
Immunofluorescence Analysis
Cells seeded on cover glasses in 12-well plates were incubated following the same protocol as described earlier. The cells were fixed in 4% paraformaldehyde and then treated with 0.25% Triton X-100 (Beyotime, China). After blocking, the cells were incubated overnight with a primary antibody against GPX4 (1:100). The next day, the cells were treated with a secondary antibody (1:100) and DAPI (5 μg/mL) (Beyotime, China) for 1 h and 10 min, respectively. The images were captured and analyzed using a confocal microscope system (Leica TCS SP8, Germany).
Western Blot Analysis
Cells seeded in 60-mm dishes were treated according to the previously described procedure. Total protein was extracted using RIPA lysis buffer (NCM, China), while nuclear protein was collected using a Nuclear Protein Extraction Kit (Boxbio, China). Protein concentrations were determined using the BCA Assay (Beyotime, China). Equal amounts of protein were loaded onto SDS-PAGE gels and subsequently transferred to PVDF membranes (Millipore, USA). After blocking, the membranes were incubated with the following primary antibodies: GPX4 (1:1000), Nrf2 (1:1000), HO-1 (1:1000), Lamin B1 (1:10000), and β-actin (1:10000). The membranes were then treated with secondary antibodies (1:20000). Chemiluminescent signals were captured and quantified using an ECL Chemiluminescence Kit (Vazyme, China) and the ChemiDoc XRS + Imaging System (Bio-Rad, USA). Lamin B1 or β-actin was selected as the internal control.
Statistical Analysis
The statistical analysis was performed using SPSS 23 and GraphPad Prism 8 software. The data were presented as mean ± standard deviation (SD) from 3 independent experiments. Differences between multiple groups were analyzed using 1-way analysis of variance (ANOVA), followed by Tukey's multiple comparison test. Statistical significance was considered for P-values < 0.05.
Results
QUE Improves RSL3-Induced Chondrocyte Viability
Figure 1A illustrates the chemical structure of QUE. Initially, the viability of C28/I2 cells was assessed after treatment with different doses of either QUE or RSL3 for 12, 24, or 48 h. Administration of QUE below 10 μM for either 12 or 24 h did not show a significant effect on cell viability (P > 0.05). However, treatment with QUE at doses of 25, 50, or 100 μM resulted in a decrease in cell viability (P < 0.01). Prolonged treatment with QUE (5, 10, 25, 50, or 100 μM) for 48 h significantly reduced cell viability (P < 0.05), except for the lower doses (0.1, 0.5, 1, or 2.5 μM), which had no noticeable influence on cell viability (P > 0.05) (Figure 1B). Treatment with RSL3 at concentrations of 0.1, 0.25, 0.5, 1, 2, 4, or 8 μM for 12, 24, or 48 h resulted in decreased cell viability (P < 0.01) (Figure 1C). Interestingly, treatment with QUE (0.5, 1, 2.5, 5, or 10 μM) for 24 or 48 h improved cell viability induced by RSL3 (0.5 μM) (P < 0.05) (Figure 1D). Therefore, the administration of 1 μM QUE or 0.5 μM RSL3 for 24 h was chosen for subsequent studies. The CCK-8 assay results demonstrated that QUE and Fer-1 significantly improved cell viability induced by RSL3 (P < 0.01) (Figure 1E). Furthermore, treatment with the Nrf2 agonist TBHQ significantly improved cell viability (P < 0.01), whereas treatment with the Nrf2 inhibitor ML385 further decreased cell viability induced by RSL3 (P > 0.05) (Figure 1F). Additionally, QUE (1 μM), Fer-1 (10 μM), TBHQ (10 μM), and ML385 (10 μM) alone did not show a significant effect on cell viability, as indicated by the CCK-8 assay results (P > 0.05) (Figures 1E and F).
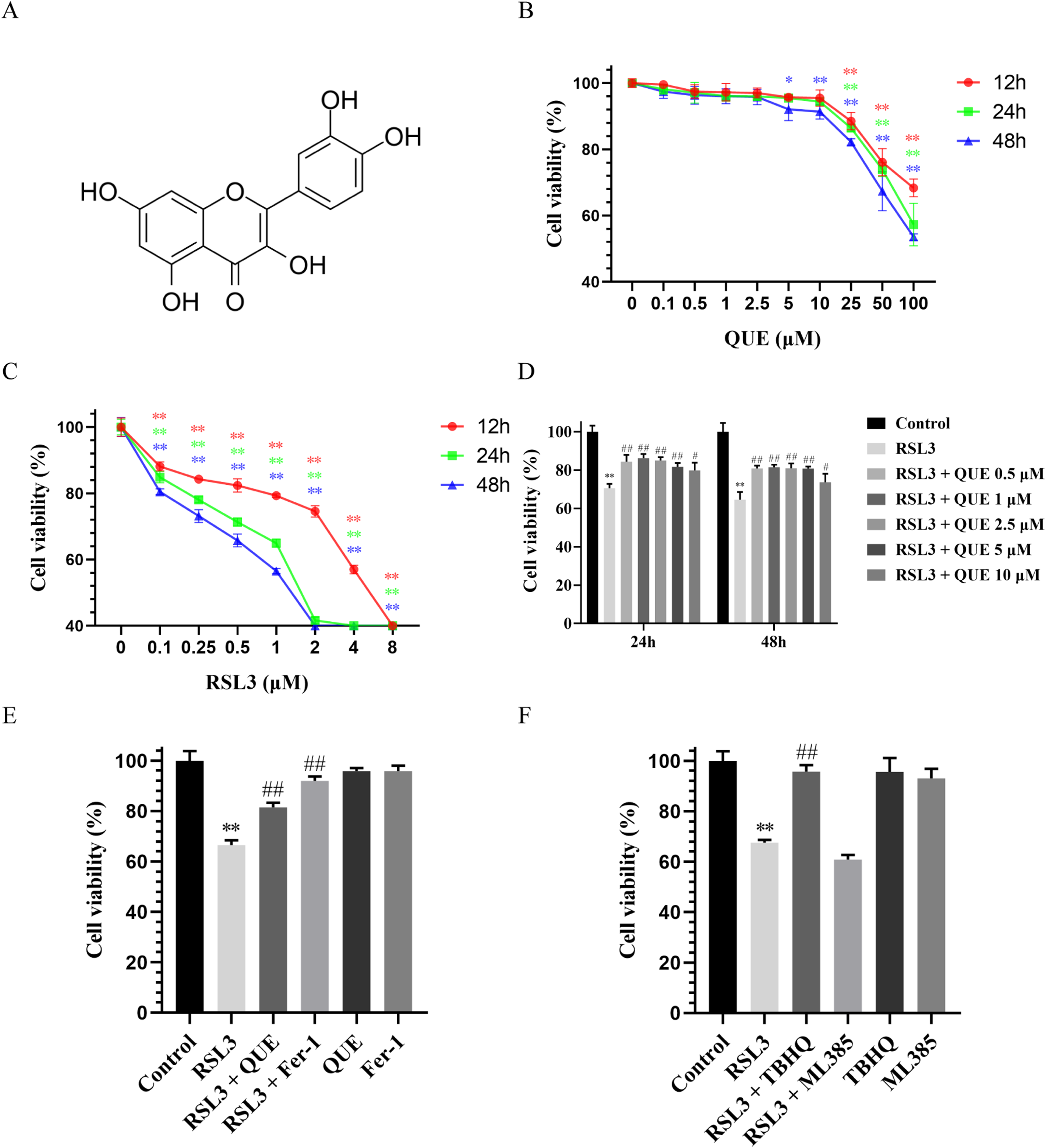
Effects of QUE, Fer-1, TBHQ, and ML385 on viability in RSL3-stimulated chondrocytes. (A) Chemical structure of QUE. (B) The effect of QUE (0.1, 0.5, 1, 2.5, 5, 10, 25, 50, 100 μM) on chondrocyte viability administered for 12, 24, or 48 h (n = 3). (C) The effect of RSL3 (0.1, 0.25, 0.5, 1, 2, 4, 8 μM) on chondrocyte viability administered for 12, 24, or 48 h (n = 3). (D) The effect of QUE (0.5, 1, 2.5, 5, 10 μM) on chondrocyte viability induced by RSL3 (0.5 μM) administered for 24 or 48 h (n = 3). (E) The effect of QUE (1 μM) or Fer-1 (10 μM) on chondrocyte viability induced by RSL3 (0.5 μM) administered for 24 h (n = 3). (F) The effect of Nrf2 agonist TBHQ (10 μM) or inhibitor ML385 (10 μM) on chondrocyte viability induced by RSL3 (0.5 μM) administered for 24 h (n = 3). * P < 0.05, ** P < 0.01 versus control; # P < 0.05, ## P < 0.01 versus RSL3.
QUE Reverses RSL3-Induced Accumulation of Lipid Peroxidation
RSL3 is utilized to directly inhibit GPX4, a critical factor in cellular lipid oxidative stress. 33 C28/12 cells were treated with RSL3 to induce ferroptosis, a process characterized by an excessive accumulation of lipid ROS. 34 To measure lipid ROS, we employed C11-BODIPY 581/591, a reliable lipophilic ROS sensor. 35 As anticipated, treatment with RSL3 resulted in a significant increase in lipid ROS accumulation (green fluorescence), as observed under a fluorescence microscope. However, co-treatment with either QUE or Fer-1 effectively reduced the green fluorescence induced by RSL3 compared to the RSL3 group (Figure 2A). Consistently, flow cytometry analysis revealed that RSL3 treatment led to a notable increase in lipid ROS accumulation (P < 0.01), while supplementation with either QUE or Fer-1 prevented RSL3-induced lipid ROS accumulation (P < 0.01) (Figure 2B and C). Additionally, MDA, a byproduct of lipid peroxidation and a positive marker of ferroptosis was measured to assess whether QUE affected the levels of oxidized lipids induced by RSL3. 36 The concentration of MDA was found to be elevated upon RSL3 treatment (P < 0.01). However, either QUE or Fer-1 intervention decreased the elevated levels of MDA induced by RSL3 (P < 0.01) (Figure 2D). Moreover, QUE and Fer-1 alone did not affect lipid peroxidation accumulation in normal C28/I2 cells. Additionally, treatment with TBHQ effectively suppressed the RSL3-induced accumulation of lipid ROS and elevation of MDA levels (P < 0.01). Conversely, treatment with ML385 resulted in increased production of lipid ROS (P < 0.01) (Figure 2E to H).

Effects of QUE, Fer-1, TBHQ, and ML385 on lipid peroxidation accumulation in RSL3-stimulated chondrocytes. (A and E) The images of C11 BODIPY 581/591 fluorescent probe staining (100× magnification). Scale bars 50 µm (n = 3). (B and F) Measurement of intracellular lipid ROS level by flow cytometry (n = 3). (C and G) Quantification of intracellular lipid ROS level (n = 3). (D and H) The intracellular level of MDA (n = 3). ** P < 0.01 versus control; ## P < 0.01 versus RSL3.
QUE Upregulates RSL3-Induced GPX4 Protein Levels
Reduced levels of GPX4 protein is typically regarded as a key characteristic of ferroptosis. Immunofluorescent staining results demonstrated a minimal presence of GPX4 (red fluorescence) induced by RSL3. Compared to the RSL3 group, treatment with either QUE or Fer-1 enhanced the red fluorescence induced by RSL3 (Figure 3A). To quantitatively assess GPX4 protein levels, a semi-quantitative evaluation was conducted through Western blot analysis. Consistent with the immunofluorescent staining findings, RSL3 induction led to GPX4 inactivation (P < 0.01), while incubation with either QUE or Fer-1 enhanced GPX4 protein levels (P < 0.05). Importantly, QUE and Fer-1 alone did not impact GPX4 protein levels in normal C28/I2 cells (Figure 3B and C). Nrf2 has been reported to regulate GPX4. 37 As expected, treatment with TBHQ significantly increased the red fluorescence and elevated GPX4 protein levels (P < 0.01) induced by RSL3. Conversely, there was no significant difference in GPX4 inactivation induced by RSL3 following ML385 treatment (Figure 3D to F).
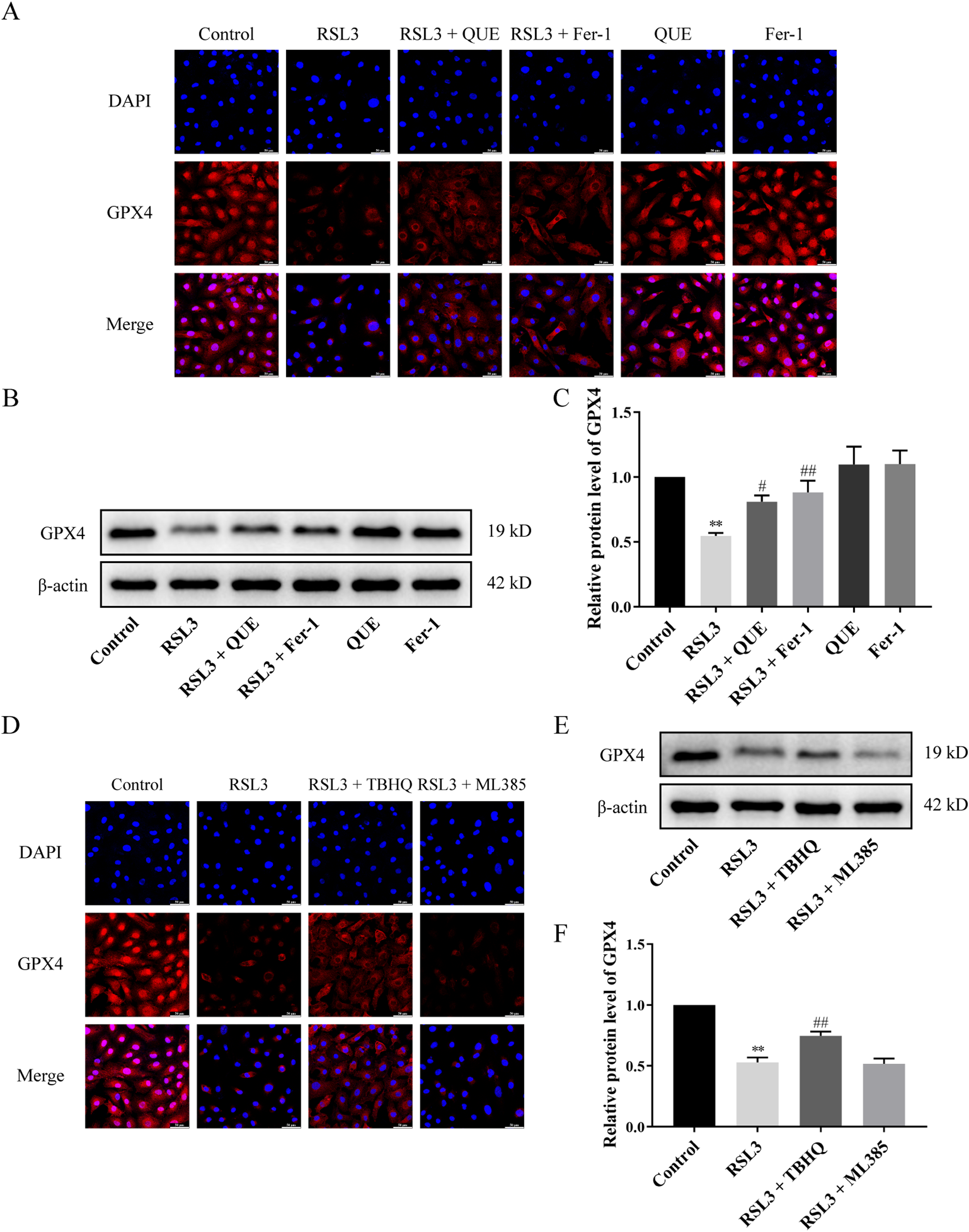
Effects of QUE, Fer-1, TBHQ, and ML385 on GPX4 protein levels in RSL3-stimulated chondrocytes. (A and D) Representative images of immunofluorescence staining (400× magnification). Scale bars 50 µm (n = 3). (B and E) Western blot validation of GPX4 (n = 3). (C and F) GPX4 protein levels with β-actin as control (n = 3). ** P < 0.01 versus control; # P < 0.05, ## P < 0.01 versus RSL3.
QUE Activates Nrf2/HO-1 Signaling Pathway in RSL3-Induced Ferroptosis in Chondrocytes
To identify crucial signals of QUE against ferroptosis in chondrocytes, the protein levels of nuclear-Nrf2 and HO-1 were examined. Treatment with RSL3 enhanced the protein levels of nuclear-Nrf2 and HO-1 (P > 0.05). Remarkably, the administration of either QUE or Fer-1 further elevated the protein levels of nuclear-Nrf2 and HO-1 (P < 0.01). Importantly, QUE and Fer-1 alone did not influence the protein levels of nuclear-Nrf2 and HO-1 in normal C28/I2 cells (Figure 4A to C). Furthermore, we employed TBHQ and ML385 to investigate the impact of the Nrf2/HO-1 pathway on RSL3-induced ferroptosis. Treatment with TBHQ significantly enhanced the protein levels of nuclear-Nrf2 and HO-1 (P < 0.01), while ML385 treatment did not reverse these changes induced by RSL3 (Figure 4D to F). These findings suggest that activation of the Nrf2/HO-1 signaling pathway mitigates ferroptosis and the anti-ferroptosis role of QUE may be dependent on the Nrf2/HO-1 signaling pathway in RSL3-stimulated chondrocytes.

Effects of QUE, Fer-1, TBHQ, and ML385 on Nrf2/HO-1 pathway in RSL3-stimulated chondrocytes. (A and D) Western blot validation of nuclear-Nrf2 and HO-1. (B and E) Nuclear-Nrf2 protein levels with Lamin B1 as control (n = 3). (C and F) HO-1 levels with β-actin as control (n = 3). ## P < 0.01 versus RSL3.
Discussion
OA is a chronic disorder influenced by various factors, such as genetics, metabolism, aging, obesity, and joint trauma.38,39 Regrettably, OA remains a challenging condition for which there are no specific therapeutic approaches available. The pathogenesis of OA is heavily influenced by chondrocytes, and chondrocyte death is the primary cause of OA. 40 In OA patients, there is a significant reduction in GPX4 protein levels in joint cartilage, and ferroptosis in chondrocytes plays a critical role in the onset and progression of OA.13,41 Ferroptosis is primarily driven by lipid peroxidation, with an excess of lipid ROS being considered as a characteristic feature of this process. 42 So, in this study, a ferroptosis model in chondrocytes was established using RSL3 stimulation to simulate the process of ferroptosis. Given the critical role of ferroptosis in chondrocytes in the pathogenesis of OA, finding suitable intervention drugs is urgently needed.
Traditional Chinese medicine has a history of thousands of years in the treatment of OA, and it is a research trend to extract its natural active monomer components to treat different diseases. QUE, a widely studied flavonoid compound, has emerged as a promising candidate for OA therapies. Numerous investigations have demonstrated that QUE exhibits significant antiarthritic and joint-protective actions through antioxidation, anti-inflammation, immunomodulation, and the promotion of cartilage repair both in vivo and in vitro.43–45 Notably, QUE has also exhibited potential in blocking ferroptosis. 46 However, research on the chondroprotective effects of QUE through the prevention of ferroptosis processes is currently limited. In this study, a ferroptosis cell model was induced by RSL3, an inhibitor of GPX4, which triggers ferroptosis by hindering its activity following induced lipid ROS accumulation and MDA production. 47 As expected, RSL3 had detrimental effects on chondrocytes and successfully induced ferroptosis in chondrocytes. On the other hand, Fer-1, as a lipophilic antioxidant, prevents membrane lipid damage by inhibiting ferroptosis through a reduction reaction. 48 Conversely, Fer-1 exhibited the opposite effect on RSL3-stimulated ferroptosis in chondrocytes. Similarly, we found that QUE exhibited beneficial effects on chondrocytes induced by RSL3, including increased cell viability, enhanced protein levels of GPX4, decreased accumulation of lipid ROS, and reduced MDA levels, suggesting that QUE holds promise as an ingredient in combating ferroptosis in chondrocytes.
QUE has been shown to promote nuclear translocation of Nrf2 and exert antioxidative stress activity.49–51 However, the relationship between QUE, Nrf2, and ferroptosis in a clinical setting has received limited attention. Nrf2 is a critical transcription factor responsible for regulating cellular redox balance. 52 Normally, Nrf2 remains inactive in the cytosol through binding to Kelch-like ECH-associated protein 1 (Keap1), which leads to its ubiquitination and subsequent proteasomal degradation. 53 However, under conditions of oxidative stress, Nrf2 dissociates from Keap1 and translocates into the nucleus. Once in the nucleus, it binds to the antioxidant response element (ARE), thereby enhancing the protein levels of GPX4 and HO-1. This process plays a vital role in maintaining the antioxidative/oxidative equilibrium.54,55 Nrf2 not only acts as a primary antioxidant transcription factor but also serves as a crucial protective factor in mitigating ferroptosis. 56 Inhibition of ferroptosis through pharmacological regulation of the Nrf2/HO-1 signaling pathway is a potential therapeutic strategy. In the current study, QUE further enhanced the activation of the Nrf2/HO-1 pathway in RSL3-stimulated chondrocytes. Interestingly, RSL3 reduced the protein levels of GPX4 while increasing the nuclear translocation of Nrf2 in chondrocytes, consistent with previous research. 57 The increased nuclear translocation of Nrf2 after RSL3 stimulation may be an intracellular self-protective response, but not sufficient to reverse ferroptosis. Surprisingly, QUE further augmented the nuclear translocation of Nrf2 in RSL3-treated chondrocytes, whose effect is to induce antioxidant enzyme gene expression, as demonstrated by the enhanced protein levels of GPX4 and HO-1, thus reversing ferroptosis by reducing the accumulation of lipid peroxidation. Furthermore, the Nrf2 agonist TBHQ inhibited RSL3-induced ferroptosis in chondrocytes by promoting the activation of the Nrf2/HO-1 pathway, leading to increased cell viability, enhanced GPX4 protein levels, reduced accumulation of lipid ROS, and decreased levels of MDA. In contrast, the Nrf2 inhibitor ML385 showed no beneficial effect on RSL3-induced ferroptosis in chondrocytes. These findings confirm the critical role of the Nrf2-mediated antioxidant system in determining the fate of ferroptosis in chondrocytes. QUE protects against RSL3-induced ferroptosis in chondrocytes by triggering the activation of the Nrf2/GPX4 signaling pathway.
However, there are limitations to consider, as the present study focused solely on a ferroptosis cell model using RSL3. Future research should explore the mechanism of QUE in ferroptosis cell models induced by erastin or other inducers, as well as investigate the anti-ferroptosis and chondroprotective effects of QUE in animal models of OA. This will contribute to the elucidation of the therapeutic efficacy of QUE in OA and provide valuable insights for clinical drug development.
Conclusions
In summary, the results of this study demonstrate that QUE effectively inhibits RSL3-induced ferroptosis in chondrocytes through the activation of the Nrf2/GPX4 signaling pathway, thereby presenting a promising option for OA treatment (Figure 5). These findings not only emphasize the significant impact of QUE on features of ferroptosis in chondrocytes but also lay the groundwork for future experimental investigations on QUE and its therapeutic potential.
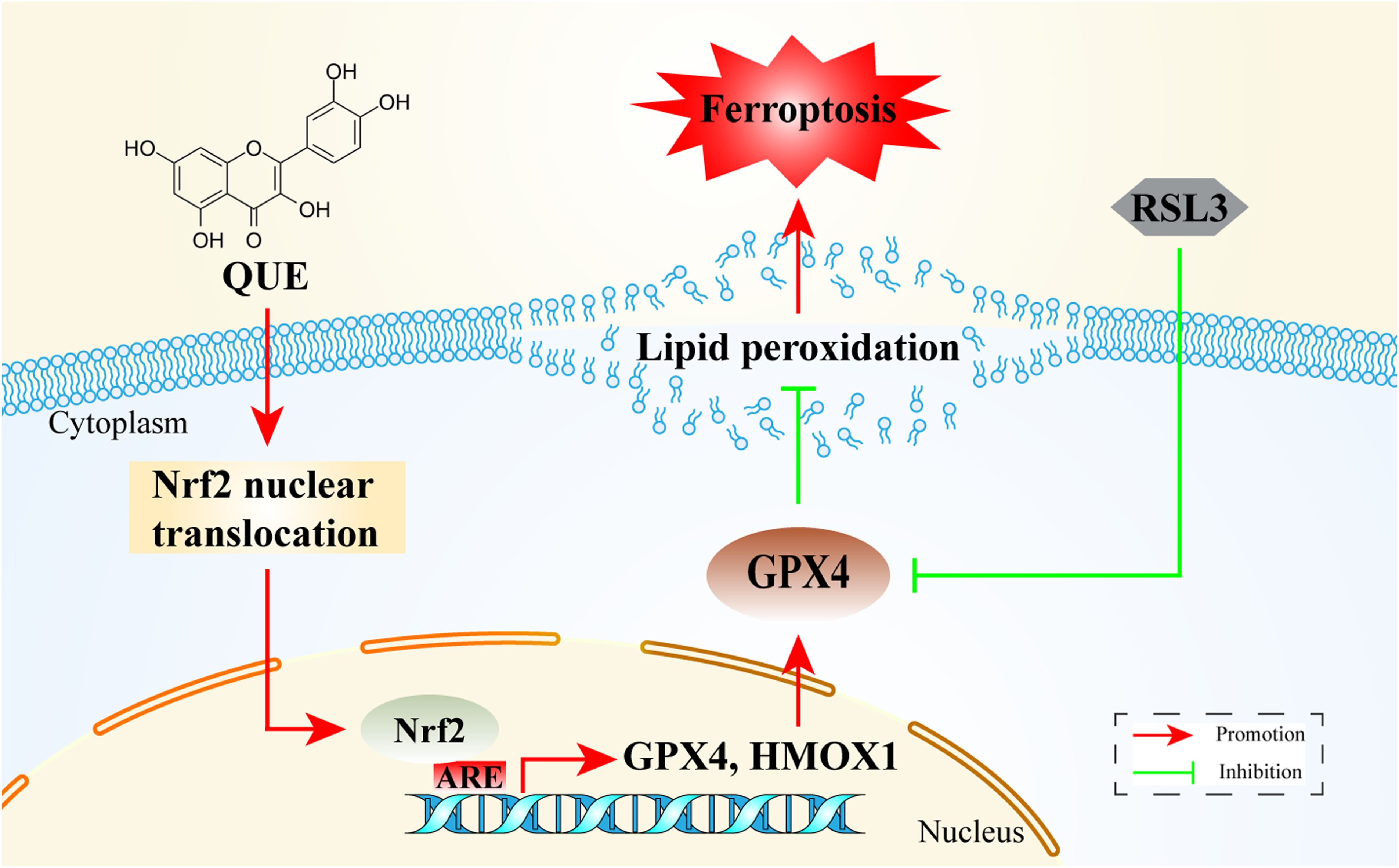
A mechanistic overview of the suppression of ferroptosis in RSL3-induced chondrocytes by QUE. QUE improved cell viability, reduced the accumulation of lipid ROS, decreased MDA levels, and suppressed ferroptosis in chondrocytes by activating the Nrf2/GPX4 signaling pathway.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X231194837 - Supplemental material for Quercetin Suppresses Ferroptosis in Chondrocytes via Activating the Nrf2/ GPX4 Signaling Pathway
Supplemental material, sj-docx-1-npx-10.1177_1934578X231194837 for Quercetin Suppresses Ferroptosis in Chondrocytes via Activating the Nrf2/ GPX4 Signaling Pathway by Gengrui Xu, Minghao Lu, Liang Fang, Fengxiang Tian, Huaxu Zhu, Lingling Zhou and Xueping Zhou in Natural Product Communications
Footnotes
Author Contributions
XPZ, LLZ, and HXZ conceived and designed the study. GRX and MHL performed experiments, prepared figures, and wrote the manuscript. FXT processed and analyzed experimental data. LF instructed experimental procedures and revision of manuscripts. All authors read and approved the manuscript.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the School-Enterprise Cooperation Projects of Clinical Approval, Patent Technology Transfer and New Drug Research and Development of Gubi Granules (contract number (2020) ZC-YY-062) and the Postgraduate Research and Practice Innovation Program of Jiangsu Province (grant number SJCX22_0753).
Statement of Human and Animal Rights
Not applicable.
Statement of Informed Consent
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.