
Abstract
Jiedu Huoxue Decoction (JHD), a recommended traditional prescription for patients with severe COVID-19, has appeared in the treatment protocols in China. Based on bioinformatics and computational chemistry methods, including molecular docking, molecular dynamics (MD) simulation, and Molecular Mechanics Generalized Born Surface Area (MM/GBSA) calculation, we aimed to reveal the mechanism of JHD in treating severe COVID-19. The compounds in JHD were obtained and screened on TCMSP, SwissADME, and ADMETLab platforms. The compound targets were obtained from TCMSP and STITCH, while COVID-19 targets were obtained from Genecards and NCBI. The protein-protein interaction network was constructed by using STRING. Gene Ontology (GO) and KEGG enrichment were performed with ClueGO and R language. AutoDock vina was employed for molecular docking. 100 ns MD simulation of the optimal docking complex was carried out with AmberTools 20. A total of 84 compounds and 29 potential targets of JHD for COVID-19 were collected. The key phytochemicals included quercetin, luteolin, β-sitosterol, puerarin, stigmasterol, kaempferol, and wogonin, which could regulate the immune system. The hub genes included IL6, IL10, VEGFA, IL1B, CCL2, HMOX1, DPP4, and ACE2. ACE2 and DPP4 were related to SARS-CoV-2 entering cells. GO and KEGG analysis showed that JHD could intervene in cytokine storm and endothelial proliferation and migration related to thrombosis. The molecular docking, 100 ns MD simulation, and MM/GBSA calculation confirmed that targets enriched in the COVID-19 pathway had high affinities with related compounds, and the conformations of the puerarin-ACE2, quercetin-EGFR, luteolin-EGFR, and quercetin-IL1B complexes were stable. In a word, JHD could treat COVID-19 by intervening in cytokine storm, thrombosis, and the entry of SARS-CoV-2, while regulating the immune system. These mechanisms were consistent with JHD's therapeutic concept of “detoxification” and “promoting blood circulation and removing blood stasis” in treating COVID-19. The research provides a theoretical basis for the development and application of JHD.
Keywords
Introduction
Coronavirus disease 2019 (COVID-19) has spread worldwide, with more than 6 million deaths. SAR-CoV-2 variants constantly appear while the vaccine's effectiveness is not so reliable. Since the COVID-19 outbreak, various traditional herbal medicines from different countries have been used to treat infected patients with promising results.1–3 As a traditional Chinese medicine (TCM), Jiedu Huoxue Decoction (JHD, 解毒活血汤) has been recommended in treatment protocols for severe COVID-19 by the National Health Commission of China and provincial health agencies. 4 It was founded by Qingren Wang (王清任) in China's Qing Dynasty. It consists of Forsythiae Fructus, Radix Puerariae, Radix Bupleuri, Aurantii Fructus, Angelicae Sinensis Radix, Radix Paeoniae Rubra, Rehmanniae Radix, Carthami Flos, Persicae Semen, and Licorice. JHD combined the efficacy of “detoxification”(解毒), “promoting blood circulation and removing blood stasis”(活血). In the Qing Dynasty, it played an essential role in treating cholera and plague. 5
Thrombosis is common in patients with severe COVID-19, with complications such as pulmonary infection and multiple organs injuries. 6 Blood stasis is a concept of Chinese medicine, and its pathogenesis in modern medicine may be endothelial injury and thrombosis. 7 The TCM therapy of promoting blood circulation and removing blood stasis based on heat-clearing and detoxification has been applied to treat severe infectious with blood stasis. TCM doctors believed that this therapy should be used in the treatment of COVID-19. 8 JHD might resist not only severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, but also inhibit thrombosis.
The aggravation of many infectious diseases is related to cytokine storms. 9 Cytokine storm was essential in transforming COVID-19 from light to severe and critical. 10 Severe inflammation caused by high levels of cytokines, which resulted in damage to the immune, blood, respiratory, gastrointestinal, and other organ systems. 11 Thus, in addition to directly inhibiting viral replication, it is also vital to reduce inflammation to avoid the cytokine storm. We speculated that the inhibition of the cytokine storm might be a factor in its curative effect on major epidemic diseases in the Qing Dynasty.
Although JHD was officially recommended as a Chinese herbal prescription for severe COVID-19, its potential bioactive substances and molecular mechanisms were unclear. Network pharmacology or bioinformatics is consistent with the overall concept of TCM, which can systematically reveal the relationship among active compounds, target proteins, and pathways. It has been used to explore the mechanism of treating COVID-19 with some traditional Chinese medicinal compounds.12–14 This study aimed to investigate the material basis and molecular mechanism of JHD in treating severe COVID-19 by in-silico methods, including bioinformatics, molecular docking, and molecular dynamics (MD) simulation.
Results
Active Compounds Preliminarily Screened
According to criteria bioavailability (OB) ≥ 30% and drug-likeness (DL) ≥ 0.18, 90, 29, 23, 22, 21, 17, 5, 5, 3, and 2 active compounds were preliminarily screened from Licorice, Radix Paeoniae Rubra, Persicae Semen, Carthami Flos, Forsythiae Fructus, Radix Bupleuri, Radix Puerariae, Aurantii Fructus, Rehmanniae Radix, and Angelicae Sinensis Radix, respectively. Then, 190 active compounds were obtained by combination and de-duplication. Notably, puerarin was a potential drug for the treatment of COVID-19. 15 It significantly affected the binding of the spike protein of SARS-COV-2 with ACE2 in human cells. 16 In addition, puerarin (OB = 24%, DL = 0.69) in TCM was usually refined into an injection. 17 Therefore, although puerarin failed to meet the criteria, it was still reserved for follow-up studies.
The Potential Target of JHD in Treating COVID-19
Four hundred and fifty-six protein targets were retrieved from the TCM systems pharmacy database and analysis platform (TCMSP) and STITCH databases, related to 147 of 190 compounds preliminarily screened. 323 and 74 COVID-19 targets were retrieved from the Genecards and NCBI databases. After merging and eliminating duplication, 378 COVID-19 related targets were achieved. They were mapped to the compound targets, and 29 common targets were obtained, which were considered the potential targets of JHD in treating COVID-19 (Figure 1). ACE2 was an essential receptor for SARS-COV-2 to enter the human body. It has been proved that SARS-COV-2 infects human beings by binding to ACE2 on cell membranes and entering cells. 18 Thus, ACE2 was given a key test in our research.

The Venn diagram of compound targets and COVID-19 related targets.
Compounds Further Screened and Herb-Compound-Target (H-C-T) Network Analysis
By comparing the data of the corresponding relationship between compounds and targets, 95 compounds were identified to interact with potential therapeutic targets. More absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of these compounds, except OB and DL, were evaluated before the H-C-T network construction. We checked them for blood-brain barrier (BBB) permeability, P-glycoprotein substrate, and adverse drug reactions (ADRs), including human Ether-a-go-go Related-Gene (hERG) blockers, human hepatotoxicity (H-HT), and Ames toxicity. Eighty-four compounds were reserved for the construction of the H-C-T network, excluding those whose ADRs were evaluated as “ + + + ” (Supplemental Table S1). The results showed that most compounds had no severe ADR. Further screening of compounds did not lead to the reduction of potential targets involved in network construction.
The H-C-T network had 123 nodes and 291 edges. These nodes included 10 herbs, 84 compounds, and 29 potential targets (Figure 2). Among 13 compounds with a degree value above 3 (Table 1), only β-sitosterol, sitosterol and bicuculline were assessed as “ + + ” in hERG blockers, indicating that they may have moderate hERG toxicity. The hERG Blockers, H-HT, and Ames toxicity of other compounds were all good. Formononetin, isoformononetin, and β-carotene were predicated to have a BBB permeability effect, which indicated that they might have potential intervention effects on nervous system infection in Covid-19. Naringenin was predicted to be a P-glycoprotein substrate. Nodes with degrees larger than twice the median were considered key nodes (Table 2). The key herbs were Licorice, Carthami Flos, and Forsythiae Fructus. The key compounds were quercetin, luteolin, β-sitosterol, puerarin, stigmasterol, kaempferol, wogonin, formononetin, baicalein, and sitosterol. Quercetin was derived from Licorice, Forsythiae Fructus, Carthami Flos, and Radix Bupleuri, acting on 16 targets. Luteolin was derived from Forsythiae Fructus and Carthami Flos, acting on 10 targets. The key compounds might be the main active pharmaceutical ingredients in JHD for treating COVID-19. The key targets were AR, DPP4, PRKACA, VEGFA, HMOX1, SREBF2, IL6, CCL2, and IL10. They might be important targets of JHD for COVID-19. The H-C-T network showed that one compound might act on multiple targets, and multiple compounds might also act on the same target. This coincided with JHD's pharmacological effects on COVID-19 through a multi-component and multi-target way.

Herb-compound-target network. The green triangle nodes represent all the herbs in JHD, sky blue square nodes represent compounds, and purple circular nodes represent targets. The larger the node, the greater degree value.
Information on Some Active Compounds Acting on Potential Targets of COVID-19.

The prediction probability values are represented with different symbols: 0-0.1( − − −), 0.1-0.3( − −), 0.3-0.5( − ), 0.5-0.7( + ), 0.7-0.9( + +), and 0.9-1.0( + + + ). Usually, the token ‘ + + + ’ or ‘ + +’ represents the molecule is more likely to be toxic or defective, while ‘ − −’ or ‘ − ’ represents nontoxic or appropriate.
Abbreviations: BBB, blood-brain barrier permeant; Pgp-sub, P-gp substrate; hERG, hERG blockers; H-HT, human hepatotoxicity; AMES, AMES toxicity; RB, Radix Bupleuri; CF, Carthami Flos; LC, Licorice; FF, Forsythiae Fructus; RP, Radix Puerariae; ASR, Angelicae Sinensis Radix; RPR, Radix Paeoniae Rubra; PS, Persicae Semen; RR, Rehmanniae Radix; AF, Aurantii Fructus.
Information on Key Nodes in the H-C-T Network.

Protein-Protein Interaction (PPI) Network Analysis
The 29 potential therapeutic targets were imported into the String database. The PPI network data from String was further analyzed and visualized in Cytoscape (Figure 3). The proteins isolated in the network were removed. In the network, there were 24 nodes and 77 edges. A node represented a protein, and an edge represented an interaction between proteins. IL6, IL10, VEGFA, IL1B, and CCL2 were the top 5 targets with the highest degrees, IL6, VEGFA, HMOX1, IL10, and IL1B had the highest betweenness centrality, and DPP4, ACE2, IL6, IL10, and VEGFA had the highest closeness centrality. The union of the above targets contained IL6, IL10, VEGFA, IL1B, CCL2, HMOX1, DPP4, and ACE2, which were considered hub targets.

PPI network. The larger the node, the greater the degree value, and the thicker the edge, the larger the combined score. The light blue column inside the node represents closeness centrality and the light red column represents betweenness centrality. The higher the column, the greater the value.
Gene Ontology (GO) Enrichment Analysis
There were 45, 7, and 10 terms enriched in Biological Process (GO-BP) (Figure 4A), Molecular Function (GO-MF) (Figure 4B), and Immune System Process (GO-ISP) (Figure 4C), respectively. According to the results of GO enrichment, JHD was mainly involved in the regulation of endothelial cell proliferation, regulation of blood vessel endothelial cell migration, positive regulation of lymphocyte activation, positive regulation of chemokine production, chemoattractant activity, cellular response to lipopolysaccharide, and response to molecules of bacterial origin and other biological processes. The molecular function of JHD in treating COVID-19 was mainly reflected in cytokine activity, type I interferon receptor binding, dipeptidyl-peptidase activity, chemoattractant activity, and lipoprotein particle binding. The enrichment results of GO-ISP showed that JHD mainly participated in T cell activation involved in immune response, B cell proliferation, CD4-positive, and alpha-beta T cell activation. This indicated that the active ingredients of JHD could play a role in multiple ways.

The visualization analysis of GO enrichment analysis. (A) GO-BP; (B) GO-MF; (C) GO-ISP.
Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment and Herb-Compound-Target-Pathway (H-C-T-P) Analysis
Fifty-seven KEGG pathways were enriched, and 16 targets were involved. The top 20 pathways (Figure 5) were ranked by P value, and the 3 pathways with the highest P value were cytokine-cytokine receptor interaction, human cytomegalovirus infection, and Coronavirus disease-COVID-19. Other pathways mainly included the following categories: (1) immune system pathways, including IL-17 signaling pathway, Toll-like receptor signaling pathway, and cytosolic DNA-sensing pathway. (2) Infectious disease-related pathways, including influenza A, tuberculosis, Yersinia infection, and malaria. (3) Signal transduction pathways, including the JAK-STAT signaling pathway, TNF signaling pathway, and HIF-1 signaling pathway. The H-C-T-P (Figure 6) network did not include other disease pathways outside COVID-19. The Coronavirus disease-COVID-19 pathway involved the 7 herbs in JHD (Radix Bupleuri, Radix Puerariae, Radix Paeoniae Rubra, Carthami Flos, Licorice, Forsythiae Fructus, and Aurantii Fructus). The compounds involved in this pathway included puerarin, quercetin, wogonin, naringenin, luteolin, and paeoniflorin. The targets participating in this pathway were ACE2, IL6, EGFR, IL1B, CCL2 (MCP-1), CXCL10 (IP-10), IFNB1, and IFNA1. ACE2 was involved in SARS-CoV-2 entering cells in the pathway, and IL6, IL1B, CCL2, and CXCL10 were cytokine storm-related targets (Figure 7).

The KEGG pathway enrichment analysis of potential targets.

Herb-compound-target-disease-pathway network. It does not include disease pathways other than COVID-19. The V-shape nodes represent all the herbs in JHD, square nodes represent compounds, circular nodes represent targets, and diamond nodes represent pathways. The nodes with red border represent Coronavirus disease-COVID-19 pathway (hsa05171) and its associated targets, compounds, and herbs. The red edges represent the interaction between the nodes with red border.

A part of the coronavirus disease-COVID-19 (hsa05171) pathway. The red nodes in the figure represent potential therapeutic targets involved in the pathway.
Results of Molecular Docking
We used the molecular docking method to evaluate the binding energy between the enriched targets (ACE2, IL6, EGFR, IL1B, CCL2, CXCL10, IFNB1, and IFNA1) in the Coronavirus disease-COVID-19 pathway (hsa05171) and their interacting compounds (puerarin, quercetin, wogonin, naringenin, luteolin, and paeoniflorin) in the H-C-T-P network. The grid boxes for docking centered on the geometrical centers of the co-crystallized ligand binding spheres present in crystals19–23 of IL6, EGFR, IL1B, IFNB1, and CCL2. The central coordinates of the CXCL10, and IFNA1 crystals24,25 with no co-crystallized ligand served as those of the docking boxes (Table 3). In this study, puerarin, as an ACE2 inhibitor, was docked with the high-resolution crystal of ACE2 (PBD ID: 1R42).26,27 We set up the docking box of puerarin and ACE2 at the binding pocket of ACE2 inhibitors, reported in our previous research 28 and other literature. 26 This box centered just on the geometrical centers of the ACE2 crystal and was big enough to accommodate the active sites, including residues Tyr510, Arg514, Asn394, and Glu398 in the pocket mentioned above.
Grid Box Coordinates and Size Parameters Used in AutoDock Vina.

These protein crystals have no co-crystallized ligand, and their macromolecule centers served as the docking box centers.
Table 4 shows that the affinity energies of all compounds with their targets were all less than −5.0 kcal/mol, indicating high binding affinities between them. 29 Among them, puerarin and ACE2 had the highest affinity, with the lowest binding energy −8.2 kcal/mol. The binding energies for quercetin docking to EGFR and IL1B were−7.3 and −7.0 kcal/mol, respectively. The binding energy between luteolin and EGFR was −7.0 kcal/mol.
The Binding Energy and Interaction Force of Compounds and Corresponding Targets Enriched in the Coronavirus Disease-COVID-19 Pathway.

The optimal binding conformations of puerarin, quercetin, and luteolin with the targets showed tight binding patterns in the active pocket (Figure 8A-D). These compounds were bonded to receptor proteins mainly through Van der Waals force, conventional hydrogen bonds, and hydrophobic interactions, including alkyl and pi-alkyl interactions. Generally, receptors and ligands formed stable complexes relying on hydrogen bonds, hydrophobic interactions, and Van der Waals forces between ligands and residues in the binding site.30,31 Figure 8A shows that puerarin formed hydrogen bonds with residues ASP350, TYR385, and PHE390 of ACE2. It was further stabilized in the binding pocket through hydrophobic interactions with PHE40 and PHE390. Puerarin also interacted with ACE2 via Asn394, Asp350, and His378, the same binding sites as a reported ACE2 inhibitor. 28 On EGFR, quercetin formed hydrogen bonds with AGR841, ASN842, and MET793, and hydrophobic interaction with VAL726, ALA743, LEU844, and LEU718 (Figure 8B). Figure 8C shows that Arg841, Met790, and Met793 of EGFR form hydrogen bonds with luteolin.

The represented results for the binding mode of compounds with targets. (A) 3D and 2D binding models of puerarin with ACE2 (PDB ID: 1R42); (B) 3D and 2D binding models of quercetin with EGFR (PDB ID: 6LUD); (C) 3D and 2D binding models of luteolin with EGFR (PDB ID: 6LUD); (D) 3D and 2D binding models of quercetin with IL1B (PDB ID: 5R85).
The Results of MD Simulation and Molecular Mechanics Generalized Born Surface Area (MM/GBSA) Calculation
The 4 complexes’ narrow root mean square deviation (RMSD) fluctuations indicated that the small molecules were tightly bonded with proteins and the complexes were stable (Figure 9). The puerarin-ACE2 complex system reached equilibrium after a short time, with slight fluctuations at 30 to 50 ns and around 90 ns. The whole system was relatively stable, and the protein backbone fluctuated in the field of 1.5 to 2.0 Å. The quercetin-EGFR complex RMSD reached a plateau in 30 ns. RMSD of the luteolin-EGFR complex system reached its peak at 50 ns, and then showed a regression trend, reaching equilibrium at 70 ns. RMSD of the quercetin-IL1B complex system rose slowly, reaching a plateau at 40 ns, and the protein backbone fluctuated in the range of 1.0 to 1.5 Å.
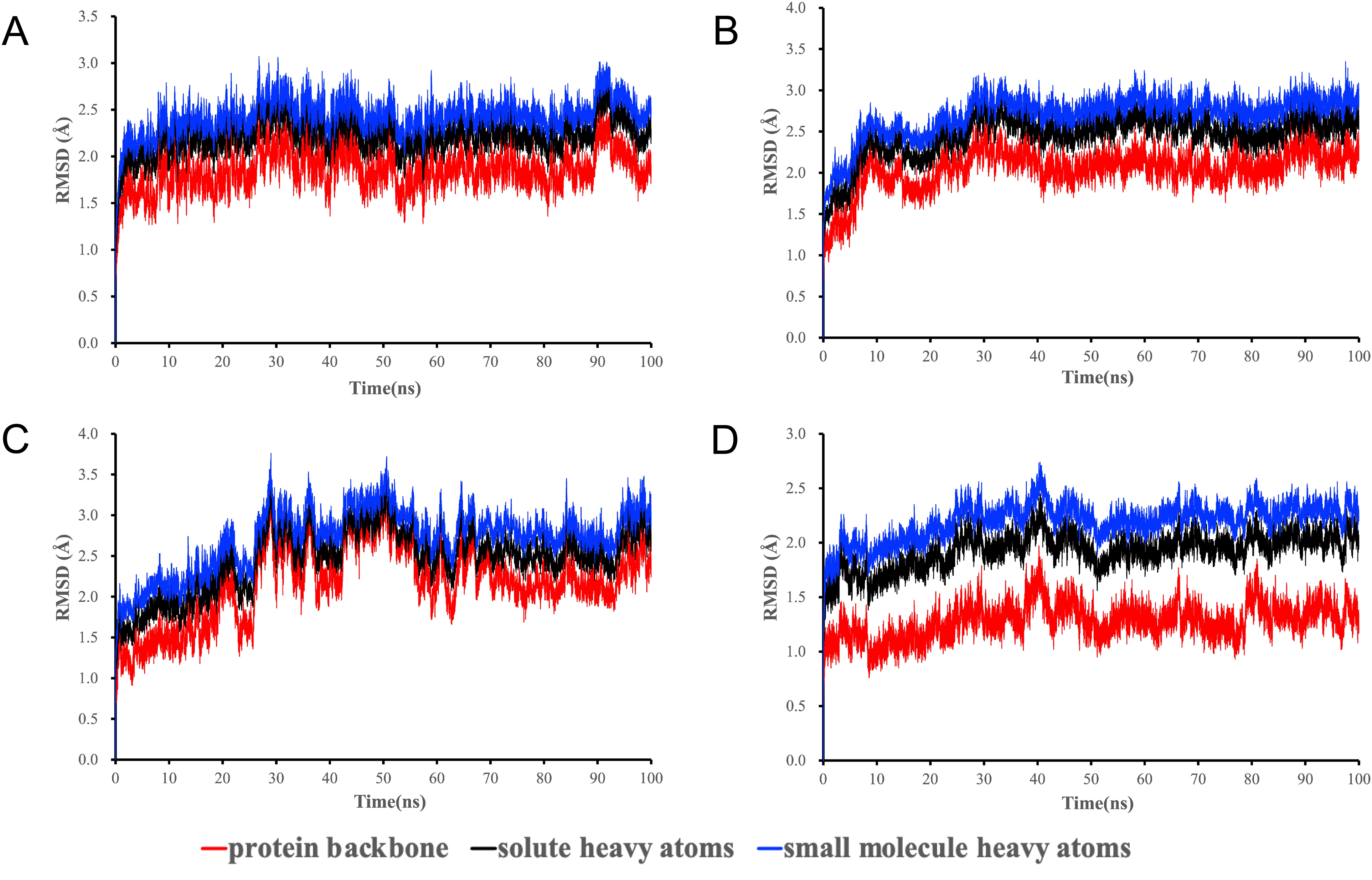
RMSD fluctuation of the protein backbone, solute heavy atoms, and small molecule heavy atoms in complexes during molecular dynamics simulation. (A) RMSD of ACE2-puerarin; (B) RMSD of EGFR-quercetin; (C) RMSD of EGFR-luteolin; (D) RMSD of IL1B-quercetin.
The MD simulation and MM/GBSA calculation indicated that all these phytochemicals could form stable complexes with corresponding targets receptors. The total binding free energy (Table 5) of the quercetin-EGFR complex was −30.8450 kcal/mol, which was the lowest among the 4 complex systems, resulting in the strongest binding affinity. The results showed that the Van der Waals force was the main force of quercetin and the EGFR complex system. Van der Waals force also played an important role in combining luteolin with EGFR, and quercetin with IL1B. The total binding free energy of puerarin and ACE2 complex was −16.5162 kcal/mol. The electrostatic interaction was far greater than non-polar solvation interaction and Van der Waals force. Electrostatic action was more conducive to the combination of puerarin and the ACE2 complex than Van der Waals force.
Molecular Mechanics Generalized Born Surface Area (MM/GBSA) Binding Free Energy.

Abbreviations: Delta G vdw, Van der Waals energy term; Delta G ele, electrostatic energy term; Delta G polar, polar solvation energy; Delta G nonpolar, nonpolar solvation energy; Delta G gas, molecular mechanics term (energy in gas phase); Delta G solv, solvation energy; Delta G total, total binding free energy.
Discussion
Our study found that the main active compounds were flavonoids and their derivatives, which might regulate host immune function.32,33 Among them, quercetin from Licorice, Forsythiae Fructus, Carthami Flos, and Radix Bupleuri in JHD had anti-inflammatory and antiviral activities,34,35 and was a potential anti-COVID-19 compound.36,37 Besides, naringenin was regarded as a candidate against SARS-CoV-2 infection. 38 Kaempferol could repair acute lung injury via the MAPK signaling pathway. 39 Both β-sitosterol and stigmasterol could alleviate the inflammatory reaction induced by LPS in the macrophage model. 40 HMGB1 was located at the core of the GO-BP network (Figure 4A). HMGB1 belonged to the damage-related molecular model and was a potential target protein for COVID-19 therapy. 41 It is usually released by damaged lung cells. 42 When serum HMGB1 increased, the inflammation of COVID-19 patients infected would persist. The H-C-T network showed that wogonin was the only related compound acting on HMGB1. Luteolin could reduce the release of HMGB1 by destabilizing c-Jun and inhibit the inflammation cascade induced by HMGB1 by lowering the Akt protein. 43 Thus, we inferred that wogonin and luteolin has the potential to intervene in COVID-19 infection by acting on HMGB1. After SARS-CoV-2 had infected the host, type I interferon decreased to a very low level, 44 and the innate immune response was seriously disturbed. 45 Moreover, GO-ISP (Figure 4C) analysis showed that IFNB1, IFNA1, and HMGB1 activated T cells and the proliferation of B cells and played a role in alleviating lymphopenia, from which most COVID-19 patients suffered. 46 The H-C-T network showed that puerarin was the only related compound acting on IFNB1 and IFNA1. Given this, we speculated that puerarin could protect against SARS-CoV-2 infection by regulating the levels of IFNB 1 and IFNA 1.
Bacterial co-infection and secondary bacterial infection were vital risk factors for the severity and mortality of COVID-19. 47 KEGG analysis showed that JHD might resist microbial infection and alleviate the deterioration of COVID-19 48 by regulating the Toll-like receptor signaling pathway, IL-17 signaling pathway, and various infectious disease pathways. Besides, gastrointestinal infections such as diarrhea and vomiting were other main symptoms of COVID-19 patients. 49 Chinese medicine believes that “lung and large intestine are interior-exteriorly related” (肺与大肠相表里). It was recorded in “Yilin Correction” (医林改错) that JHD was influential in the treatment of diarrhea and vomiting in patients with Vibrio cholera infection. 50 Our study showed that the targets IL6, IL10, IL1B, IFNG, and IL4 of JHD were enriched in the inflammatory bowel disease (hsa05321) pathway. It might be the mechanism of JHD intervention on intestinal infection in severe COVID-19 patients.
This study showed that quercetin, luteolin, naringin, paeoniflorin, and wogonin in JHD could act on storm-related targets IL6, IL1B, CCL2, CXCL10, and EGFR enriched in the Coronavirus disease-COVID-19 pathway (hsa05171). Cytokines play an essential role in the pathophysiology process of COVID-19. 51 Therefore, in addition to inhibiting virus replication, it is crucial to avoid cytokine storms. Molecular docking results showed that the above-mentioned compounds and their corresponding targets had high binding activity. The MD simulation and MM/GBSA calculations of the EGFR-quercetin complex, EGFR-luteolin complex, and IL1B-quercetin complex showed stability during 100 ns. IL6, as a hub gene with the largest degree value in the PPI network, was a clinical predictor of disease severity in COVID-19. 52 IL6 was an essential factor in inducing cytokine storm. 53 The release of pro-inflammatory factors such as IL6, IL1B, and TNF in inflammation could aggravate inflammation and tissue damage. 54 Chemokines CCL2 and CXCL10 could regulate the activation and migration of immune cells when stimulated by antigens. 55 Anti-inflammatory factors IL10 and IL4 in the PPI network strongly interacted with pro-inflammatory factors IL6 and IL1B. They could reduce the inflammatory reaction, and limit the bioavailability of pro-inflammatory factors. 56 EGFR could be activated by acute lung injury and lead to phosphorylation of STAT3, 57 thus regulating the level of circulating IL6. 58 PPI network analysis showed that JHD might intervene in the cytokine storm by adjusting the balance among pro-inflammatory factors, anti-inflammatory factors, and chemokines to avoid the aggravation of the disease.
Clinical studies 6 found that thrombosis was the most important reason for the deterioration of COVID-19 patients besides cytokine storm. Venous thrombosis was common in severe COVID-19 patients, especially pulmonary embolism, and even disseminated intravascular coagulation. Early detection and effective prevention of thrombosis would reduce the mortality rate. 59 Endothelial injury is an essential factor for thrombosis in COVID-19. 60 GO-BP analysis showed that VEGFA, PRKACA, HMOX1, HMGB1, APOE, and CCL2 regulated the proliferation and migration of endothelial cells. Elevated VEGFA level in serum of COVID-19 patients was crucial to endothelial homeostasis in blood vessels. 61 PRKACA could regulate the activity of forkhead box protein O1 (FoxO1) in vascular endothelial cell migration. 62 HMOX1 could inhibit platelet aggregation and had antithrombotic properties. 63 In addition, ACE2 might be a negative regulator of ANGII biological activity, improving endothelial dysfunction. 64 We speculated that JHD might influence the proliferation and migration of endothelial cells, thereby intervening in thrombosis by regulating VEGFA, PRKACA, HMOX 1, HMGB 1, APOE, and CCL2.
At present, the drug targets against SARS-CoV-2 infection mainly include human protein ACE2, transmembrane protease serine 2 (TMPRSS2), and various SARS-CoV-2 proteins such as spike protein (S-protein), 3C-like protease (3CLpro), and papain-like protease (PLpro).65,66 ACE2 is a crucial receptor for SARS-CoV-2 to invade the human body. 18 Small molecular compounds acting on ACE2 as inhibitors can block the binding of SARS-CoV-2 to ACE2. In this study, puerarin, as an ACE2 inhibitor, was docked with ACE2. The results of molecular docking, MD simulation, and MM/GBSA calculations showed that puerarin had high binding activity and could form stable complexes with ACE2, which was consistent with the conclusion of Qin. 15 Both hydrogen bonds and hydrophobic interactions might stabilize puerarin at the binding site of ACE2 and enhance the binding affinity and drug efficacy. 67 It was also found that puerarin could regulate the ACE2 levels in the kidney, heart, and other organs. 68 Therefore, we could conclude that puerarin might prevent the virus from entering the human body by inhibiting ACE2 and blocking the binding of SARS-CoV-2 to ACE2. In addition, DPP4 might be another critical target of SARS-CoV-2, having a high affinity for the SARS-CoV-2 spike protein. 69 Inhibition of DPP4 might reduce the virus's entry into the respiratory tract and replication in COVID-19 patients. 70 There were 36 active compounds in the H-C-T network (Table 2) that could bind to DPP4, such as luteolin, kaempferol, wogonin, and baicalein. Transmembrane protease serine 2 (TMPRSS2) triggering the spike protein of SARS-CoV-2 is the first biological step in infection. Androgen receptor (AR) activity is necessary for transcription of the TMPRSS2 gene. 71 This was consistent with the report that males with a high-level AR were more likely to develop severe COVID-19. 72 There were 70 compounds in JHD that could act on AR (Table 2). Above all, we considered that JHD could intervene in the entry of SARS-CoV-2 into the host by regulating ACE2, DPP4, and AR.
Conclusions
JHD could treat COVID-19 systematically through multiple targets and multiple pathways. We speculate that JHD could treat COVID-19 by interfering with cytokine storm, thrombosis, and SARS-CoV-2 entering cells and regulating the host immune system. These mechanisms were consistent with the treatment concept of “detoxification” and “promoting blood circulation and removing blood stasis” in TCM for COVID-19. We believe that JHD should be paid more attention to and applied as an auxiliary medicine for the heat-clearing and detoxicating TCMs such as Lianhua Qingwen Capsule recommended in COVID-19 Traditional Chinese Medicine Diagnosis and Treatment Plan. Given the limitations of bioinformatics analysis and computational chemistry methods, 73 including the inability of public databases to update the information in time, the conclusions of this study inevitably have some defects. Pharmacological mechanisms of JHD active ingredient need to be further verified by in vivo and in vitro experiments.
Materials and Methods
Preliminary Screening of Compounds of JHD
All the compounds of the 10 herbs in JHD were obtained from the TCMSP database (https://tcmspw.com/tcmsp.php). The compounds were preliminarily screened according to the criterion of OB ≥ 30% and DL ≥ 0.18.74,75 The screened active compounds were submitted to the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), and their structures and names were confirmed and standardized.
Retrieving Targets of the Compounds Preliminary Screened
TCMSP and STITCH databases (http://stitch.embl.de/) were used to obtain the action targets of the active compounds preliminarily screened above. UniProt website (http://www.uniprot.org/) was used to standardize the names of proteins and corresponding genes. The compounds with no target were removed at this step.
Potential Therapeutic Targets for COVID-19 of JHD
After entering the keywords “COVID-19” and “Coronavirus disease 2019” into the Genecards (https://www.genecards.org/) and the NCBI (https://www.ncbi.nlm.nih.gov/gene/) databases, all the “Homo sapiens” genes related to COVID-19 were collected. We mapped the JHD target with the COVID-19 related genes to obtain common targets, which were considered potential targets of JHD in treating COVID-19. After the above work, the compounds unrelated to these potential therapeutic targets were excluded.
Further Screening of Compounds and Construction of the H-C-T Network
The SWISSADME database (http://www.swissadme.ch/) was employed to examine the BBB permeability, and P-glycoprotein substrate of compounds reserved in the previous steps. In addition, ADMETLab 2.0 database (https://admetmesh.scbdd.com/) was used to examine the ADRs, including hERG Blockers, H-HT, and Ames Toxicity. The compounds evaluated as “ + + + ” in ADRs were eliminated. Cytoscape (version 3.7.2) 76 was used to construct the H-C-T network, which visualized the relationships among herbs, the finally screened compounds, and potential therapeutic targets. Network Analyzer plug-in of Cytoscape was used to calculate the degree values of the nodes. The nodes with degrees greater than twice the median were considered important. Nodes of the herbs, compounds, and targets were sorted by degree and distributed in a circle.
Construction of PPI Network
A PPI network can provide a reference for screening hub targets. The potential targets of JHD in treating COVID-19 were submitted to STRING (version 10.5, https://string-db.org/) to construct a PPI network with a confidence of 0.70. Cytoscape was used to visualize the PPI network. Network analyzer plug-in was used to calculate network parameters, including degree value, betweenness centrality, and closeness centrality. Those nodes with high values of the 3 parameters were considered hub nodes.
GO Enrichment Analysis
To understand further the function of the target genes, ClueGO (version 2.5.6) and CluePedia (version 1.5.7) plug-in77,78 of Cytoscape were used to perform GO enrichment analysis of potential therapeutic targets in GO-BP, GO-MF, and GO-ISP. We used a two-sided hypergeometric statistical test. The cut-off P values for GO enrichment in GO-BP, GO-MF, and GO-ISP were 0.01, 0.05, and 0.05, respectively. The functional groups were created by the iterative merging of initially defined groups based on the predefined kappa score threshold of 0.4. The final groups were randomly colored and overlaid with the network. The label for the most significant term in every functional group was in bold font and the same color as the group. The red labeled nodes represent the target genes, and the other nodes represent the GO terms. The larger the GO node, the lower the P value.
KEGG Pathway Enrichment Analysis and H-C-T-P Network Construction
R language (version 3.6.1) and its packages clusterProfiler 79 and pathview 80 were used to perform the KEGG pathway enrichment analysis on the potential therapeutic targets. Functional enrichment analysis was based on the P value ≤ .05. The analysis results are presented in the form of bubble diagrams. Cytoscape was used to construct the H-C-T-P network, connecting the critical pathways and the corresponding therapeutic targets. Nodes of the herbs, compounds, and targets were distributed circularly.
Compound-Target Molecular Docking
We used AutoDock vina (version 1.1.2) 29 for molecular docking to verify the binding activity of compounds to targets enriched in the critical pathway. The 3D structure SDF files of the compounds were downloaded from PubChem and converted into PDB files by openbabel GUI. 81 These PDB format compounds were then imported into MGLTools (version 1.5.6). 82 Their polar hydrogens were added, partial charges were calculated, flexible bonds were set to be rotatable, and finally saved as PDBQT files. The protein crystal files were downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/). The following were the selection criteria for the X-ray protein crystals: source from Homo sapiens; (2) the resolution was smaller than 2.5 angstroms; (3) the presence of one co-crystallized ligand was given priority; and (4) the PDB deposition data. Protein crystals were introduced into PyMOL (version 2.0, Schrödinger, LLC.). Their solvents, ligands, and other non-protein molecules (such as ions, metals, and cofactors) were removed and saved as PDB format files. Polar hydrogens were then added using MGLTools software, and Gasteiger charges were calculated and saved as PDBQT files. For the proteins with co-crystallized ligand, the docking grid box centered on the geometrical center of the co-crystallized ligand binding sphere obtained by Discovery Studio (BIOVIA, San Diego, CA, USA). The crystal centers served as those of the grid boxes for the proteins with no co-crystallized ligand. The box could contain the entire active pocket. The grid size was set to suitable points with a grid spacing of 0.375 Å and exhaustiveness of 9 (Table 3). During the docking procedure, the receptors were held rigid, while the ligands were allowed to be flexible.
After docking, the docking result log and the ligand conformation files in PDBQT format were outputted. The figures of 2D and 3D binding modes between active site residues and the conformation of the ligand with the lowest affinity energy were generated using the Discovery Studio.
MD Simulations
The protein-ligand complexes with the lowest binding energy obtained from the above docking process were used as the initial configuration for the MD simulation. The MD simulations were performed in the Yinfo Cloud Computing Platform (YCCP) (https://cloud.yinfotek.com) using AmberTools 20 (AMBER 2020, University of California, San Francisco). AMBER ff19SB 83 and GAFF 84 force fields were used for proteins and ligands, respectively. Using the OPC water model with a margin of 10 Å, the system was solved by a truncated octahedron water box. Counter ions (Na + and Cl-) were added to neutralize the system, and periodic boundary conditions were employed. Structural optimization was performed sequentially: (a) 2500 steps of steepest descent and 2500 steps of conjugate gradient, under a harmonic constraint of 10.0 kcal/(mol·Å2) on heavy atoms. (b) 10 000 steps of steepest descent minimization and 10 000 steps of conjugate gradient minimization. Then, the system was equilibrated during 200 and 1000 ps NPT ensembles. The temperature was maintained at 300 K using the Berendsen thermostat with 1 ps coupling constant and a pressure of 1 atm using Monte Carlo barostat with 1 ps relaxation. Finally, 100 ns MD simulations in the Canonical Ensemble (NVT) without restraint were performed. The RMSD was calculated using the CPPTRAJ module. 85
MM/GBSA Calculations
Two hundred snapshots were extracted from the MD trajectory and applied for MM/GBSA
86
binding free energy (“Δ” G_bind) calculation according to the following equation:
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221096966 - Supplemental material for Jiedu Huoxue Decoction for Cytokine Storm and Thrombosis in Severe COVID-19: A Combined Bioinformatics and Computational Chemistry Approach
Supplemental material, sj-docx-1-npx-10.1177_1934578X221096966 for Jiedu Huoxue Decoction for Cytokine Storm and Thrombosis in Severe COVID-19: A Combined Bioinformatics and Computational Chemistry Approach by Ying Liu, Han Yan, Hui-bin Jia, Li Pan, Jia-zheng Liu, Ya-wen Zhang, Jing Wang, Dao-gang Qin, Lei Ma and Ting Wang in Natural Product Communications
Footnotes
Author Contributions
Conceived and designed the study: Ying Liu. Bioinformatics analysis: Ying Liu, Li Pan, Jing Wang, Ting Wang. Computational chemical analysis: Ying Liu, Jia-zheng Liu, Ya-wen Zhang. Pharmacology and pathology discussion: Ying Liu, Li Pan, Lei Ma, Dao-gang Qin. Wrote or contributed to revising the manuscript: Ying Liu, Ya-wen Zhang, Ting Wang. All authors have read and approved this version of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Traditional Chinese Medicine Science and Technology Development Program of Shandong Province, Natural Science Foundation of Shandong Province, Medicine and Health Science Technology Development Program of Shandong Province (grant number 2019-0890, 2020Q128, ZR2017PH054, 2017WS327).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.