
Abstract
Drugs are continuously being evaluated for novel therapeutic uses. The purpose of this work was to screen anticancer triazole/tetrazole derivatives for effectiveness against the SARS-CoV-2 main protease (Mpro). First, the chemical structures’ activity was derived from conceptual quantum chemical calculations. According to molecular docking analysis, the compounds scored good interactions against SAR-COV-2's Mpro, with binding energies extending from −8.21 to −8.97 kcal/mol. The docked complexes included various bindings with His41 and Cys145, both catalytic residues responsible for cleavage of the SARS-CoV-2 Mpro. Among the 4 studied compounds, TD3 exhibited the highest affinity by achieving the most stable binding energy and lowest value for the inhibition constant. Most striking was that TD3 not only formed strong bonds with the catalytic residues His41 and Cys145, but also captured the residues of the catalytic loop (Cys44 to Pro52), which flank the catalytic dyads in Mpro's active site. As a result, the studied triazole/tetrazole derivatives, notably TD3, must be reviewed as potent drugs that could be repurposed for SARS-CoV-2 treatment.
Introduction
SARS-CoV-2 continues to be a public health emergency that has captivated the entire world's attention. Yet, there is no ultimate efficient therapy for SARS-CoV-2, justifying the massive research efforts geared toward developing safe and effective drugs. Pandemics, like COVID-19, are characterized by sudden expansion, and rapid escalation of health and economic sequences, with very huge stresses imposed on both. Unfortunately, drug development processes are usually time intensive with thorough evaluations for safety and efficacy being needed for drug approval. Hence, drug repurposing/repositioning is considered useful, cost-effective, and time-saving for discovering new treatments.
Drug repurposing has been widely employed to discover new clinical opportunities for known drugs.1,3 It has been proposed for the adoption of a number of drugs in the treatment of SARS-CoV-2.4,7 Some of the drugs are commercially available and well known,8,9 while others are based on preclinical studies.10,11 In both cases, the repurposing/repositioning method is faster and less expensive than the novel synthesis. 12
With virus-infected cells and cancer cells showing similar features, the treatment goals for both SARS-CoV-2 and cancer are considered similar.13,14 Hence, therapeutic goals include reducing inflammation, inhibiting cell growth and division, and modulating the host's microenvironment. Indeed, notable numbers of medications reconsidered for SARS-CoV-2 therapy are anti-cancer medications. Accordingly, many researchers have exhaustively reviewed the potential effectiveness of cancer drugs against SARS-CoV-2 infection.15,16
Computational chemistry is a rapidly growing field that depends on the efficiency of computers and advanced algorithms to predict and interpret molecular systems. The development of quantum chemical techniques requires the formulation of many chemical and mathematical concepts to provide accurate methods for calculating the properties of chemical systems. 17 Density functional theory (DFT) is widely regarded as one of the most effective methods for describing the ground-state properties of different types of materials. The success and popularity of the DFT method in the field of computational chemistry can be attributed to its high accuracy in describing small/large molecules over the course of many prior publications.18,21 Molecular docking is a computational method for evaluating ligand–protein interactions with the help of virtual screening software. The applicability of the docking algorithm for virtual screening was proven by the accuracy of simulating the experimental binding pose. Molecular docking aids in the discovery of drugs that can modify the protein's biological activity, as well as providing scientific data on potential therapeutic targets. 22 There have been numerous works devoted to the molecular docking approaches to search for potential SARS-CoV-2 main protease (Mpro) inhibitors using natural or synthetic molecules from various chemical libraries.23,28
In this paper, DFT and molecular docking methodologies are used to explore the inhibition capability of screened anticancer triazole–tetrazole derivatives against the Mpro of the SARS-CoV-2 virus. First, DFT calculations are used to characterize the selected compounds in terms of some reactivity parameters. Molecular docking is next employed to investigate and analyze the binding affinity, binding modes, and stability of interactions inside the active site of the SARS-CoV-2 Mpro.
Materials and Methods
Experimental Details
A new 1, 2, 3-triazolyl, along with a combination of triazole–tetrazole molecules, were synthesized and screened as antibacterial/ anticancer drugs by Pal et al. 29 The design depends on linking more than one pharmacophoric group through linkers to yield hybrid molecules containing biologically active components, resulting in highly efficient pharmacological agents. IR, 1H NMR, and 13C NMR spectroscopy, as well as mass spectrometry, was used to analyze the structure of the synthesized compounds. The toxicity risks, and druglikeness of all the chosen compounds were examined and their antibacterial and anticancer activities as oral, bioavailable drugs were screened on the basis of Lipinski's “Rule of five”. The results demonstrated that most of the proposed molecules have significant therapeutic potential, including activity against human colon and lung cancer cells. Specifically, four of the studied molecules were more cytotoxic than the known adriamycin against all examined human cancer cell lines; 2 of the 4 compounds have triazole structures, while the other 2 are triazole–tetrazole hybrids.
This research aims to repurpose these four compounds and establish effectiveness against SARS-CoV-2. Table 1 shows the chemical formula and molecular structure of the selected compounds, which have been referred to as TD1, TD2, TD3, and TD4; each an acronym for first, second, third, or fourth triazole/tetrazole derivative, respectively. Several studies confirmed Mpro as the virus's key enzyme, in view of its essential role in viral transcription and replication. Hence, Mpro is an attractive therapeutic target for SARS-CoV-2, and as a result, many studies conducted on its inhibition.30,33 In this study, Mpro was used as the target protein in molecular docking simulations.
Chemical Formula, Molecular Structure, and Optimized Structure for the Selected Compounds.

Computational Methods
Gaussian 09W software 34 was used to calculate the optimum molecular structure for the ground state of the mentioned molecules. The calculations were carried out using the DFT method by applying the theoretical model Beck's three-parameter exchange function in combination with Lee–Yang–Parr nonlocal correlation functional (B3LYP) and 6-311G** basis set. 35 The optimized structures, highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) surfaces, and molecular electrostatic potential (MEP) patterns were visualized using the graphical interface code Gauss View 6. 36
The molecular docking procedure was performed using Autodock tools 37 to estimate the binding energies and identify the main binding sites of the docked complexes (SARS-CoV-2 Mpro and the aforementioned molecules). The Mpro of coronavirus was obtained from the Protein Data Bank (PDB code: 6lu7), then prepared by removing water molecules, adding the polar hydrogen atoms, and assigning the Gasteiger charges to all atoms of the protein. The ligand was positioned in a grid box in the Mpro protein at coordinates x = 11.824, y = 14.735, and z = 74.152, with grid size fixed to 40 × 40 × 40 xyz points and 0.375 Å grid spacing. The optimized structures of DFT calculations were used as the initial ligands structures for the docking simulation. The Lamarckian genetic algorithm (LGA) has been used to execute the docking calculations with 100 runs for each process and the other parameters were maintained at their default values. The docking results have been analyzed and visualized by Discovery Studio Visualizer software. 38
Results and Discussion
DFT Calculations
HOMO and LUMO Analysis
The HOMO and the LUMO, commonly known as the frontier molecular orbitals, are massively important descriptors for the electronic structures of compounds. The ability to donate electrons defines HOMO, whereas the ability to accept electrons characterizes LUMO. The patterns of the frontier molecular orbital surfaces for the selected ligands are shown in Figure 1. HOMO orbitals of TD1 were provided by the 2-naphtholanion group and the next CH2 methylene on the left side of the molecule, whereas LUMO is distributed on the right side over the 2H-chromen-2-one plus the adjacent oxygen atom. Due to structural similarity, the HOMO and LUMO patterns of the first and second molecule are almost identical, except TD2 has a small LUMO lobe on the H atom in the methyl group. The contribution in HOMO for TD3 comes from the 1-phenyl-1H-tetrazole, followed by the sulfur atom and methylene group, and LUMO is delocalized over 2-chloro-6-methylquinoline, excluding the methyl group. The HOMO orbitals of TD4 are extremely similar to those in the TD3 molecule with a larger contribution of sulfur atoms. LUMO orbitals, on the other hand, are uniformly dispersed over the nitrobenzene group.

The patterns of frontier molecular orbital of the molecules under study.
Quantum Chemical Descriptors
To link molecular properties with the chemical reactivity of molecules, some popular chemical notions must be determined. These chemical notions are known as quantum chemical parameters such as band gap energy (ΔE), chemical hardness (η), chemical softness (σ), electronegativity (χ), and electrophilicity (ω). The following equations are used to determine the mentioned reactivity descriptors using Koopman's theorem:
39


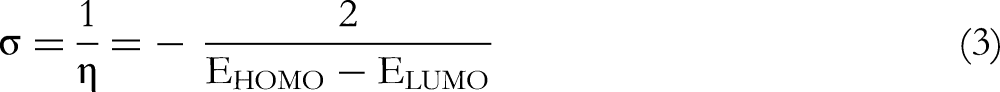


Calculated Quantum Chemical Quantities in the Form of HOMO and LUMO Energies, Energy gap, Chemical Hardness, Chemical Softness, Electronegativity, and Electrophilicity index.

Note: HOMO = highest occupied molecular orbital; LUMO = lowest occupied molecular orbital.
Molecular Electrostatic Potential
One of the most important metrics in investigating and interpreting molecular reactivity is the MEP. MEP is a map of electrical charge clouds due to the distribution of electrons and nuclei, and is used to forecast the regions where electrophilic and nucleophilic reactions may happen within the molecule. MEP is displayed in color code to indicate the regions of the molecule with neutral, positive, and negative electrostatic potential. A favorable site for electrophilic attack is represented by the red area (negative potential), a favorable site for nucleophilic attack by the blue area (positive potential), and a zero potential area by green.
For TD1 and TD2 structures, the most negative regions were observed around two nitrogen atoms of the triazole group, oxygen of the 2-naphtholanion group, and on the carbonyl of the 2H-chromen-2-one group. Positive areas were mostly found near the H-atoms bonded to the groups in the molecules’ center. TD3 has just two negative sites, one on the nitrogen atoms of the 1-phenyl-1H-tetrazole group and the other to a smallish extent on the nitrogen atoms of the triazole group. For the last derivative, the nitrogen atoms of 1-phenyl-1H-tetrazole and triazole groups, as well as the nitro group, were shown to have negative potential sites. The positive sites were localized on the hydrogen atoms of both the triazole group and nitrobenzene ring.
The importance of MEP lies in its ability to identify easily the chemical activities of molecules, through the considerable distribution and density of red and blue-colored spots. As shown clearly in Figure 2, the densest red or blue regions appeared on TD1 and TD2, then TD4 and TD3. This is compatible with the previous classification of these molecules in terms of smaller energy gap and high chemical reactivity TD1 → TD2 → TD4 → TD3. Furthermore, it is evident that TD3 is the most stable because it only has two small areas of negative potential (red color), whereas the green color (zero potential) spans the entire molecule.

Surfaces of the color-coded molecular electrostatic potential (MEP) of the studied structures.
Molecular Docking
A good binding affinity, established using molecular docking analysis, is required for a drug to be effective against SARS-CoV-2's Mpro. The lower the free binding energy, the more stable the interaction between a ligand and its protein receptor. Table 3 lists the docking binding energies, inhibition constants, the interacting amino acid residues, interaction types, and their equivalent band length. The four compounds displayed binding energies ranging from −8.21 → −8.97 kcal/mol. The achieved docking scores are noteworthy compared to previously tested drugs, with the 1, 2, 3-triazole derivatives used in this study having binding affinities predicted to be between −6.0 and −8.8 kcal/mol ligand.40,41 The least binding energy (−8.97 Kcal/mol) of TD3 qualifies it as a potent inhibitor of SARS-CoV-2 Mpro. TD1, TD3, and TD4 have only one hydrogen bond interaction with residues HIS41, GLU166, and THR26, respectively. On the other hand, TD2 was able to form 3 hydrogen bond interactions with GLU166, CYS145, and TYR54.
The Binding Affinity Results of the Selected Compounds with 6lu7 Main Protease (Mpro) of COVID-19.
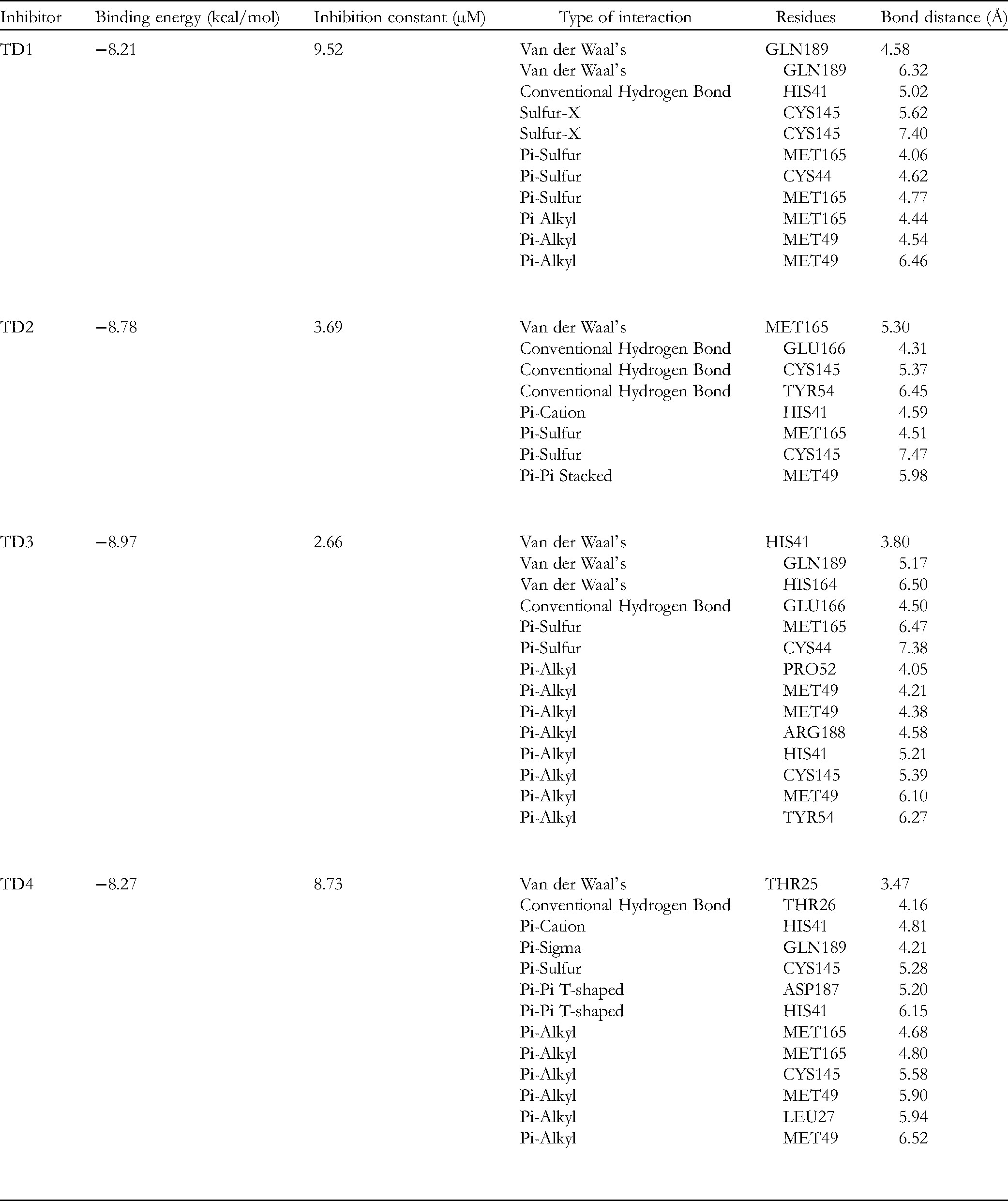
The inhibition constant (Ki) is used as an indicator for the inhibition of an enzyme, defined as the amount of the substance needed to reduce an enzyme's activity by half. The small value of the inhibition constant reflects a high binding affinity. The smallest Ki in Table 3 was achieved by TD3 (2.66 μM), signifying the highest binding affinity and the formation of the most stable complex with Mpro among the studied ligands.
Mpro has key functions in the transcription of SARS CoV-2 viruses. It is responsible for hydrolyzing large polyproteins and producing viral nonstructural proteins that have a fundamental role in viral replication. The dyads (His41and Cys145) are catalytic residues contained in the Mpro protein, while Cys145 acts as a nucleophile and His41 provides the base during the hydrolysis reaction. Thus, Mpro necessitates the catalytic dyads Cys 145–His 41 to cleave SARS-CoV-2 polyproteins.42,44 Hence, the interaction with the catalytic residues (His41and Cys145) is an attractive target for inhibition. Furthermore, it has been shown that the Cys44 to Pro52 residues constitute the catalytic loop, which forms the catalytic pocket, a loop that surrounds the cavity of the catalytic site in Mpro.45,46
The docking interactions between ligands (1-4) and Mpro pocket residues of SARS-CoV-2 are shown in Figure 3. TD1 formed 3 interactions with the catalytic dyad (His41 and Cys145); His41 has a strong hydrogen bond, and CYS 145 formed 2 Pi-sulfur bonds with the 2 rings of 2H-chromen-2-one. TD2 interacted with His41 and CYS 145 through a Pi-cation and hydrogen bond interaction. The binding mode of TD3 revealed notable interactions with the catalytic residues within the binding sites of Mpro; 2 alkyl interactions enabled the chlorine ion to capture the catalytic dyad (His41 and Cys145); the docked interactions also involved a van der Waal's bond with His4. In addition, TD3 is bound to the residues of the catalytic pocket (Cys44 to Pro52) by Pi-sulfur and Pi-alkyl interactions. TD4, on the other hand, bound to the catalytic dyad were Pi-cation and Pi-Pi-T-shaped with His41, then Pi-sulfur, Pi-alkyl with Cys145. Overall, the ability of forming various binding interactions with catalytic residues His41 and Cys145 suggest that the studied compounds have a high potential as potent Mpro inhibitors.

The docking interaction pattern of the selected compounds against the pocket domain of main protease (Mpro) protein.
In overview, the mentioned synthesized triazole and tetrazole compounds were tested for anticancer effects by Pal et al. 29 Using the MTT micro cultured tetrazolium assay and the standard drug adriamycin, the compounds had been evaluated for anticancer activity against human colon cancer cell line Colo-205 and human lung cancer cell line HOP-205. These compounds demonstrated superior inhibition of both cell lines. In the current work, the same compounds yielded an extremely positive performance when examined against Mpro of COVID-19. The chosen compounds interacted with a vital building unit in the cavity of the main protein. This was accomplished by inhibiting the main catalytic dyads responsible for SARS-CoV-2 Mpro replication, as well as the catalytic loop surrounding the cavity containing the catalytic dyads.
Conclusions
The COVID-19 pandemic has grown exponentially, yet no therapy has been ultimately effective. Many studies have recently been conducted to determine the suitability of cancer drugs for treating SARS-CoV-2 infection. In the present study, DFT calculations and molecular docking protocol were implemented on 4 anticancer triazole/tetrazole derivatives to identify possible Mpro inhibitors. The information about the structural chemistry of the proposed derivatives was initially investigated using HOMO-LUMO energies, quantum chemical descriptors, and MEP distribution patterns. The results of molecular docking revealed that the selected molecules had high binding affinity values for the viral Mpro protein. Different types of interactions formed between the ligands and the catalytic dyads (His41 and Cys145) which have a main role in the replication process of the SARS-CoV-2 Mpro. TD3 achieved remarkable results by binding both the catalytic dyads (His41 and Cys145) and the catalytic loop (Cys44 to Pro52) that surrounds the cavity containing the catalytic dyads. Hence, the anticancer triazole/tetrazole derivatives have been highlighted to possess strong inhibition activity against the Mpro.
Footnotes
Acknowledgments
This research was supported by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, Grant No. (21-13-18-061).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Imam Mohammed Ibn Saud Islamic University (grant number 21-13-18-061).
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.