
Abstract
Xanthine oxidase (XO) is a potential target for gout disease experiments on animals and humans. Using a molecular docking technique to search for anti-XO compounds from Vietnamese medicinal plants, we discovered that numerous compounds from Uvaria cordata (Dunal) Alston (Annonaceae family) showed this activity. Among these, cordauvarin A exhibited the strongest binding affinity (−8.8 kcal/mol) to XO through a binding interaction with 5 amino acids (eg Gln-1194, Ala-1079, Ser-1080, Met-1038, and Arg-912) of XO protein. Lipinski's rule of five was used to predict the druglikeness of this compound. To confirm the inhibitory activity, an in vitro assay was performed, and the results demonstrated that cordauvarin A significantly inhibited XO, with an IC50 of 124.5 ± 10.12 μM. This study reveals that cordauvarin A is a possible natural therapeutic agent for gout treatment and that this genus should be explored more extensively. However, further investigations are necessary to develop possible natural therapeutic medicines for clinical usage.
Introduction
Drug research and development is a time-consuming and difficult process. It is a sequential procedure that commences with target selection and progresses via lead discovery, experimental and preclinical trials. 1 Bioinformatics technologies can assist in the identification of drug targets by using virtual screening analyses.2,3 They are used to evaluate compounds and protein interactions, as well as to improve the properties of the molecules. In addition, the combination of computational and experimental methodologies such as chromatography (LC or GC) mass spectrometry and in vitro and in vivo investigations have been demonstrated to improve drug development success rates. 4
Xanthine oxidase (XO) is a form of xanthine oxidoreductase, an enzyme responsible for oxidizing hypoxanthine to xanthine and converting xanthine to uric acid from purine nucleotides. 5 As a result, XO activity leads to uric acid deposition in targeted tissues. Gout is caused by an abnormal increase in uric acid in the body, leading to the formation of urate crystals and their deposition as monosodium urate monohydrate crystals in the joints and the tissues around the joints. 6 These crystals can cause acute inflammation and tissue damage, which manifest as cartilage ulceration, osteomyelitis, and chronic inflammation of the synovial membrane in the long term.7,8 Globally, the incidence and prevalence of gout have doubled in the past two decades. 9 Nonsteroidal anti-inflammatory drugs (NSAIDs) (ibuprofen, aspirin, and diclofenac), selective COX-2 inhibitors (meloxicam), and corticosteroids (prednisone and prednisolone) are used to treat gout. Although they are effective, they form O2− free radicals and cause side effects, such as skin allergies, rashes, diarrhea, and fever. Allopurinol, an XO inhibitor, is the clinically preferred drug for the treatment of gout. However, the drug produces side effects, such as hypersensitivity reactions, Stevens–Johnson syndrome, hepatotoxicity, and nephrotoxicity. 10 Research has empirically evaluated herbs that can inhibit XO, revealing new treatment methods that help reduce side effects. Numerous natural compounds have been identified and reported as XO inhibitors.11–13 However, because of the lack of appropriate proof of drug–protein interactions, none of these compounds has continued to clinical trials. At the molecular level, interaction studies contribute to the understanding of the binding mechanism and therapeutic potential of potential drugs. In this study, we focused on the drug–protein interactions and the binding mechanism of natural compounds to XO.
The genus Uvaria L. (Annonaceae) comprises approximately 150 species. New natural products of various structures continue to be studied, some of which have shown promising antitumor, antibacterial, and antioxidant activities.14,15 In Vietnam, the genus Uvaria includes 15 species, distributed from Thanh Hoa province to the south. U cordata is commonly used in traditional medicine to treat rheumatism, tonic muscles, and cough. 16 Phytochemical analyses of U cordata have revealed the isolation of triterpenoids and chlorohydrins. 17 In our previous work, 8 compounds, namely cordauvarin A, 5β,6β-epoxylnusane-3β-ol, stigmasterol, taraxerol, velutinam, cyathoviridin, 10(14)-aromadendren-4-ol, and 1-pentadecanol, were isolated from the leaves of U cordata. In addition, we have reported the cytotoxicity of these compounds.18,19
The current trend is the development of functional foods derived from medicinal plants. Although U cordata has anti-inflammatory properties and is frequently used in traditional medicine, there has been no study on its potential to develop into a supportive treatment for gout. Therefore, in this study, to evaluate further the different biological activities of compounds from U cordata, the dataset that includes these eight compounds was selected for screening to identify potential compounds as potential XO inhibitors. The 3D crystal structure of mammalian xanthine oxidoreductases similar to human XO was selected for the docking protocol. To minimize the time and cost of experimental testing, certain compounds that pass the criteria of the screening process were considered for in vitro experiments to validate the virtual screening results. Finally, molecular docking simulation and absorption, distribution, metabolism, elimination, and toxicity (ADMET) studies were used to investigate the drug properties and molecular interactions between candidates and XO protein.
Results and Discussion
Virtual Screening
In our previous study, the information on eight natural compounds from U cordata was collected from previous literature.18,19 Three-dimensional structures of these compounds were constructed and used for the docking process with target bovine XO. The 3D structure of the bovine XO enzyme (PDB ID: 3NRZ) was selected for virtual screening with three chains of 1222 amino acids named A: (Thr-2-Lys-165), B: (Pro-224-Gly-528), and C: (Asp-571-Lys-1326). The active site of 3NRZ is a small cavity at the C chain. The grid box covered the active site of chain C found in previous studies (eg Glu-802, Arg-880, Ala-910, Gly-913, Phe-914, Phe-1005, Ser-1008, Phe-1009, Thr-1010, Leu-1014, and Pro-1076). 20 Hypoxanthine, the co-crystal ligand of bovine XO enzyme (PDB ID: 3NRZ) was re-docked to validate the selected protein structure and docking protocol by analyzing the root mean square deviation (RMSD) of the docking pose compared with the crystal pose. According to the results, the docking position has a similar conformation and orientation to the crystal pose with an RMSD of 1.327 Å (Figure 1). Because previous studies have found that a RMSD of less than 2.0 Å is acceptable and valuable for docking experiments, 21 this docking protocol could be utilized in this research.

Docking pose (yellow) and crystal pose (green) of hypoxanthine at the crystal structures of xanthine oxidase (XO) (PDB ID: 3NRZ).
Eight natural compounds, namely cordauvarin A, 5β,6β-epoxylnusane-3β-ol, stigmasterol, taraxerol, velutinam, cyathoviridin, 10(14)-aromadendren-4-ol, and 1-pentadecanol, were docked to the active site of bovine XO. Allopurinol is a well-known XO inhibitor that is commonly used in the treatment of gout. 22 In addition, allopurinol is a competitive inhibitor of XO. 23 Therefore, we selected allopurinol as a reference inhibitor. After docking, the results revealed a list of compounds with docking scores better than −7.1 kcal/mol, the docking score of allopurinol. To select bioactive compounds with a strong binding affinity for bovine XO, a cut-off of −7.5 kcal/mol was set. The results of these selected compounds are presented in Table 1. Among these compounds, the aromatic structure cordauvarin A surpassed this threshold with the strongest binding affinity (−8.7 kcal/mol). Therefore, cordauvarin A was selected for further experiments to demonstrate the potential of developing a new drug that inhibits XO.
Docking Results of Eight Natural Compounds Selected for Virtual Screening.

Xanthine Oxidase Assay
The results from the virtual screening process demonstrated that the aromatic compound cordauvarin A exhibited strong potential to inhibit XO. Accordingly, in vitro experimental investigations demonstrating the biological activity of this compound were performed to validate the docking results. Allopurinol was used as a positive control. Cordauvarin A exhibited inhibitory activity against XO, with an IC50 value of 124.5 ± 10.12 µM, whereas allopurinol, the positive control, had an IC50 value of 8.23 ± 1.10 µM
The experimental work matched the results of the virtual screening process. To explain further the biological inhibitory ability against XO, our study involved a molecular interaction analysis between the compound and the protein.
Molecular Docking Analysis
Cordauvarin A interacted with XO via two hydrogen bonds and three hydrophobic interactions (Figure 2, Table 2). The amino acid Ser-1080 interacted with cordauvarin A at the aromatic structure at a distance of 4.54 Å. In the chain structure of the compound, 2 oxygen atoms bound with XO via two hydrogen bonds with a minimum distance of 3.95 Å with Ala-1079 and 4.69 Å with Gln-1194, and a carbon atom was also bound to2 alkyl interactions with a minimum distance of 5.46 Å with Met-1038 and 5.17 Å with Arg-912. Four key amino acids, Glu-802, Ala-910, Gly-913, and Phe-914, were also responsible for the interaction at the active site of XO via Van der Waals interactions.
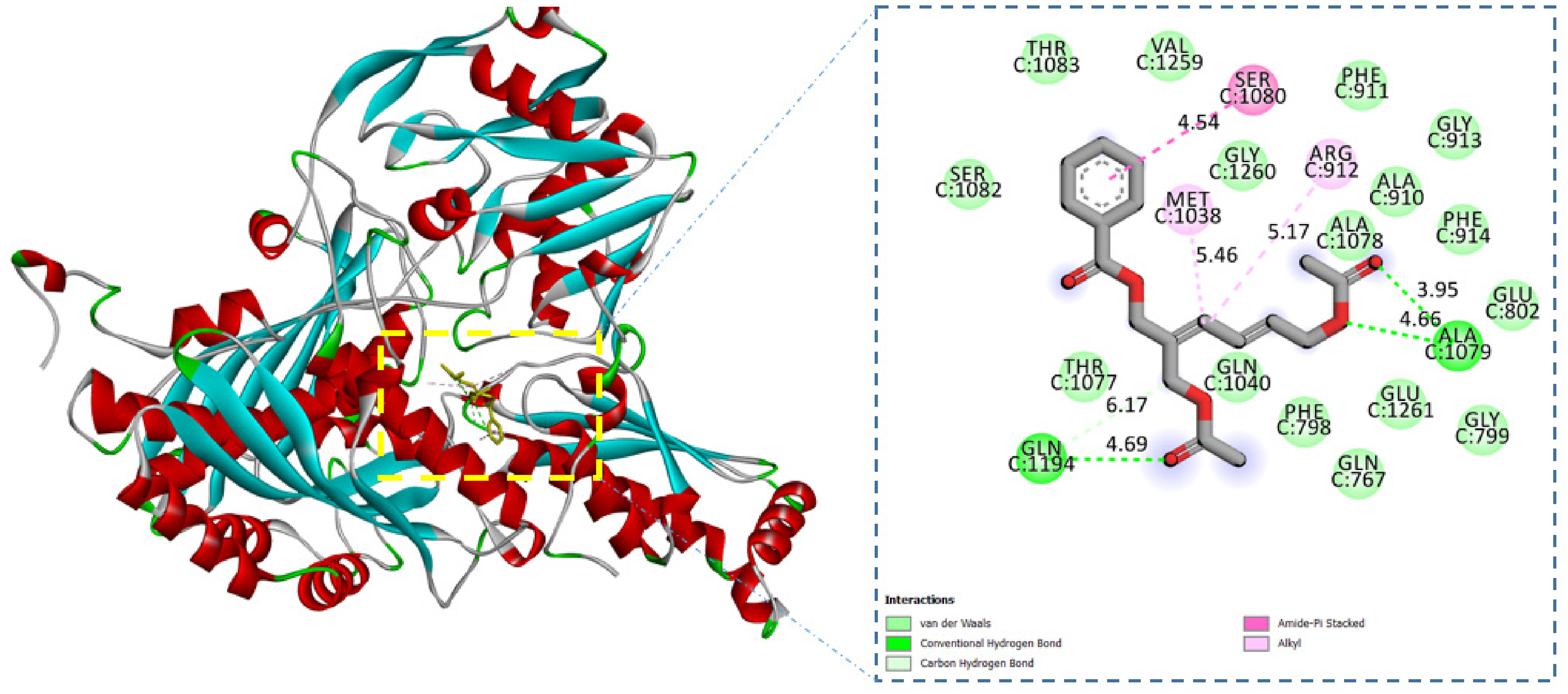
Cordauvarin A–xanthine oxidase (XO) interactions.
Docking Results of Cordauvarin A and Allopurinol Toward XO.

Abbreviation: XO: xanthine oxidase.
On the other hand, ursolic acid, febuxostat, and quercetin demonstrated activity on XO. 24 The binding pose of cordauvarin A was also a similar interaction site, which blocks the entrance gate of the channel protein. This may enhance the specificity of XO inhibitors.
The interaction between allopurinol and XO was similar to that between cordauvarin A and XO. It was highlighted that allopurinol also interacts with XO at Glu-802, Thr-1010, Arg-880, Ala-1079, Phe-1009, Phe-914 with 3 hydrogen bonds and 3 hydrophobic interactions (Figure 3, Table 2). The results showed that allopurinol has a minimum distance of 4.89 Å with Glu-802, 2.89 Å with Thr-1010, 4.34 Å with Arg-880, 4.73 Å with Arg-914, and 4.39 Å with Phe-1009. These amino acids are responsible for the interaction at the active site of XO. Moreover, two key amino acids, Ser-1008 and Leu-1014, were also responsible for the interaction at the active site of XO via Van der Waals interactions. Although the binding affinity for allopurinol was −7.1 kcal/mol, the compound forms many strong bonds and interacts with many key amino acids. This can explain allopurinol's strong inhibitory activity against XO in the in vitro experiments. In addition, cordauvarin A is considered a competitive inhibitor and a substrate for XO, which forms strong bonds at the active site through a mechanism of purine analogs.

Allopurinol–xanthine oxidase (XO) interactions.
Bovine XO is 90% similar to human XO in terms of its overall sequence. Most of the amino acids that interact with ursolic acid in human and bovine XO are conserved. 25 Notably, the amino acids at the catalytic site do not differ between human and bovine XO. Therefore, this still preserves the lipophilic character of the binding pose and their Van der Waals interaction with substance A. As a result, the recommended binding for the molecular docking simulations are relevant to the human enzyme.
ADMET Properties
To predict the characteristics of the active compound to evaluate the drug-like substance for oral use, Lipinski's rule of five was used to access the druglikeness of active compounds. The oral bioavailability of substances with good membrane permeability and hydrophobicity was demonstrated by the log P, TPSA, MW, HBA, and HBD values. The appropriate values of nRotB and MR indicated that molecules have good absorption in the intestine and high oral bioavailability.
As shown in Table 3, the results demonstrated that cordauvarin A has acceptable drug-like properties and other druglikeness parameters (MW ≤ 500 Da, log P < 5, nHBD ≤ 5, nHBA ≤ 10, and TPSA < 140 Å). 26 As a result, cordauvarin A has been proposed as a potential compound in drug development with good oral bioavailability. In addition, the TPSA value of the compound was 48.42, and the log S value was −2.97. This indicates good solubility in water. 27 From these results, the compound was predicted to have excellent absorption in the intestine. However, the nRotB value, which reflects the stereospecificity of a drug molecule, is 11. Therefore, the bonds between the compound and amino acids in space cannot be stable. Furthermore, allopurinol has no rotatable bonds; therefore, the bonds between allopurinol and XO can be more stable. This may explain the difference in biological activities of the two compounds in in vitro assays. Further studies on chemical structure are required to improve the bioactivity of this hit compound.
Drug Properties of Isolated Compounds Analyzed Using SwissADME.

Abbreviations: LogP: Log of octanol/water partition coefficient; LogS: log of solubility; MR: Molar refractivity; MW: Molecular weight; nHBA: number of hydrogen bond acceptor(s), nHBD: number of hydrogen bond donor(s); nRotB: number of rotatable bonds; TPSA: total polar surface area; XO: Xanthine oxidase.
The predictions of absorption and distribution of cordauvarin A are shown in Table 4. Cordauvarin A exhibited high gastrointestinal absorption and was predicted not to cross the blood–brain barrier. This suggests that the molecule can be developed into an oral drug with high bioavailability and no CNS toxicity. The log of skin permeability value was −6.51 cm/s. To evaluate the metabolism and excretion of cordauvarin A, the in silico SwissADME server was used to predict the interactions between cordauvarin A and cytochromes (CYPs). A variety of CYPs affect drug metabolism, with CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 predicted to be vital for drug biotransformation. 28 The results also revealed that cordauvarin A only inhibited CYP1A2 and did not inhibit either other CYPs or the substrate of permeability glycoprotein (P-gp substrate). However, the toxicity of cordauvarin A should be evaluated to ensure its safety and the potential to develop therapeutics without endangering the health of patients. In this study, to validate the toxicity of cordauvarin A, the LD50 value was expected to be 3.44 mg/kg and was classified as “non-toxic” by the DL-AOT prediction server. These findings suggested that the chemical structure of cordauvarin A is less likely to change during absorption and metabolism. Hence, cordauvarin A can achieve good bioavailability when administered orally without toxic effects and is anticipated to be safe. These results suggest that the bioactive compound identified in this study can be a potential candidate for gout treatment.
ADME Predictions of Isolated Compounds Computed by SwissADME.

Abbreviations: BBB Per: blood–brain barrier permeability; CYP, cytochrome-P; GI Abs: gastrointestinal absorption; log Kp: log of skin permeability; P-gp, permeability glycoprotein.
Conclusion
Molecular docking plays a crucial role in searching for potential plants containing bioactive compounds. This process is the first step in drug discovery and applies computational tools to reduce the time and cost to identify a new method for treating gout patients. In this study, the aromatic compound cordauvarin A exhibited the strongest binding affinity. To evaluate virtual screening results, an in vitro test was performed. Cordauvarin A extracted from U cordata showed inhibitory activity against XO with an IC50 value of 124.5 ± 10.12 μM. Molecular interaction analysis and ADMET studies were used to further explain its biological ability. These results suggested that the bioactive compound identified is a promising candidate for gout treatment. However, further in vivo and preclinical studies are needed to develop this compound into a commercial drug.
Materials and Methods
Protein and Ligand Preparation
The crystal structure of bovine XO in complex form with hypoxanthine (PDB ID: 3NRZ) was obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). Next, the crystal structures of XO were processed by removing the existing ligand and water molecules using Discovery Studio 2020. Subsequently, polar hydrogens and Kollman charges were added to the protein using AutoDockTools version 1.5.6. Finally, the macromolecule was exported into a dockable pdbqt format for molecular docking. Pymol 2.4 was used to calculate the RMSD between two molecules.
Three-dimensional structures of natural compounds from U. cordata were either retrieved from the PubChem database or prepared using MarvinSketch 20.17 (ChemAxon, USA). In the last step, all ligands were converted to dockable pdbqt format using Open Babel 3.1.1.
Molecular Docking
Virtual screening of the bioactive compounds on XO was carried out using AutoDock Vina. The grid box that covers the active site of protein was generated with the following parameters: center_x = 37.35, center_y = 19.85, center_z = 17.85, size_x = 30, size_y = 30, and size_z = 30. Docking scores are reported in kcal/mol, and the docking scores ranked the compounds. Finally, the molecular interactions between proteins and selected ligands were visualized by Discovery Studio Visualizer.
In Silico Druglikeness and Toxicity
The druglikeness of compounds was predicted using the Lipinski rule. 29 The ADME parameters of ligands were calculated using the SwissADME server, which supplied information on the ligands’ absorption, distribution, metabolism, and excretion properties. 30 The DL-AOT prediction server was used to predict acute oral toxicity (T). 31
In Vitro Assay
XO inhibitory activity was measured using the spectrophotometric method in 96-well plates, as described previously
24
, with slight modifications. In brief, the reagents were 70 nmol/L phosphate buffer pH 7.5, 150 nmol/L xanthine solution dissolved in phosphate buffer (pH 7.5), and 0.01 IU/mL XO solution (diluted from a stock enzyme solution into the buffer solution). The test samples were initially prepared in dimethyl sulfoxide (DMSO), followed by dilution with the buffer. A mixture consisting of 35 μL of phosphate buffer solution, 50 μL of working solution, and 30 μL of XO solution (0.1 units/mL) was pre-incubated at 25 °C for 15 min. The reaction only took place when 60 μL xanthine solution (750 µM) was added and incubated for 30 min at 25 °C. The reaction was stopped by adding 25 mL of HCl 1 N solution before measuring the absorbance at 290 nm on a BioTek Epoch microplate spectrophotometer. The blank of each sample was prepared in the same way, but in the absence of XO solution. The negative control (0.5% DMSO) in the absence of an inhibitor and the positive control (allopurinol) were run simultaneously. The XO inhibitory activity was measured at doses of 500, 100, 20, and 4 μg/mL and estimated as the half-maximal inhibitory concentration (IC50), calculated by the program TableCurve2Dv4. All experiments were performed in triplicate. The XO inhibitory activity was expressed as the inhibitory percentage (I) calculated using the following formula: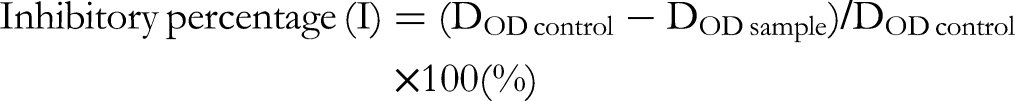
DMSO was present in the final reaction mixture at a concentration below 0.25%, indicating that this solvent had a negligible effect on XO activity.
Footnotes
Acknowledgments
This research was supported by the University of Medicine and Pharmacy, Hue University.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the University of Medicine and Pharmacy, Hue University, (grant number 22-21).
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.