
Abstract
Dendrobium fimbriatum Hook is an important medicinal and ornamental orchid that may lead to important drugs and cosmetics. In this study, 2 analytical strategies were combined to describe the detailed composition of a hydroethanolic extract of the whole plant. The CARAMEL approach, based on centrifugal partition chromatography and nuclear magnetic resonance data interpretation, allowed the rapid dereplication of 21 major constituents, mainly including polyols, nucleosides, zwitterionic compounds, phenolic compounds, and organic acids. Further semipreparative high-performance liquid chromatography and spectroscopic analyses led to the isolation of 6 additional compounds; 4 phenanthrenes (plicatol B, hircinol, plicatol A, and plicatol C), 1 bibenzyl (3′,4-dihydroxy-3,5′-dimethoxybibenzyl), and 1 furostanol saponin (protodioscin), as well as a new phenanthrene derivative, plicatol D. A total of 28 metabolites were structurally elucidated, among which 23 are described for this species for the first time.
Introduction
The Dendrobium genus, of the Orchidaceae family, contains the highest number of traditional vascular medicinal epiphytes. 1 Certainly, Dendrobium species have been used as first-rate herbs and prized folk medicine in Asia for a thousand years.2,3 They are referred to as “Habra Dendrobii shihu” in traditional Chinese medicine (TCM). 4 A recent review highlighted this genus as an important source of new drugs and cosmetics. 5 Indeed, 22 species were referenced with traditional uses in dermatological disorders, and 131 compounds isolated from Dendrobium plants have been reported to possess anti-inflammatory, antimicrobial, antioxidant, antiaging, anti-psoriasis, and tyrosinase-inhibitory activities. 5 Although Dendrobium encompasses over 1500 species, 6 only a few species, D crepidatum, D denneanum, D loddigesii, D nobile, and D officinale have been extensively studied.
The aim of this study was to assess the phytochemical components of Dendrobium fimbriatum Hook, which is an important ornamental and medicinal orchid. Dendrobium fimbriatum is found in Southeast Asia between 500 and 1500 meters of altitude. Its fringe-lipped sparkling flowers, ranging from pale yellow to deep orange, confer to the plant a high ornamental value. Dendrobium fimbriatum belongs to “Fengdou Shihu” in TCM. The stems are used for the stomach, to improve the production of saliva, and to decrease fever.
7
The leaves are reported to induce the production of body fluid. In balm form, they are applied to fracture areas to strengthen bones. The whole plant is used to treat and stabilize liver and mental disorders.
8
So far, the stems, in particular, have been phytochemically investigated (Table S1). Several compounds (
To obtain a highly detailed phytochemical profile of the whole plant, a dereplication strategy named CARAMEL (CARActérisation de MELanges, in French, for characterization of mixtures) based on centrifugal partition chromatography (CPC) and interpretation of nuclear magnetic resonance (NMR) spectroscopic analyses was carried out. 13 This identification process was complemented with semipreparative high-performance liquid chromatography (HPLC) to isolate some minor constituents and further explore the chemical diversity of the D fimbriatum extract.
Results and Discussion
CARAMEL Approach
The hydroethanolic extract of the whole plant of D fimbriatum contained metabolites from various chemical classes and exhibited a large polarity range. Therefore, a 3-phase solvent system composed of n-heptane, methyl-tert-butyl-ether, acetonitrile (ACN), and water in the proportions 1/1/1/1 (vol/vol) was selected for the fractionation process to tentatively separate a maximum of metabolites and facilitate the NMR identification process. In total, 13 final fractions were obtained in a reasonable separation time (total CPC duration of 2 h 30 min), without loss of biomass (total recovery of 97.0% for the sum of F1-F13), and, as illustrated in Figure 1, each fraction displayed a simplified chemical diversity as compared to the initial crude extract.

TLC profile and mass distribution of the 13 final centrifugal partition chromatography (CPC) fractions obtained from the aerial parts of Dendrobium fimbriatum EtOH 50% extract—(a) 254 nm; (b) 366 nm; and (c) visible after vanillin/H2SO4 reagent spraying.
After NMR analysis of all fractions, automatic peak picking, and data alignment, the resulting table was made of 13 columns corresponding to the CPC fractions and 162 rows corresponding to the chemical shift buckets (Δ 0.3 ppm) for which at least one C-13 peak was detected in at least one fraction. This table was submitted to hierarchical clustering analysis (HCA) for the recognition of similarities between the C-13 fingerprints obtained in successive CPC fractions. In this way, C-13 NMR signals belonging to the same compound were aggregated as “chemical shift clusters” in a heat map, which is given in Figure 2. An in-house database containing predicted chemical shift values of natural metabolites, including n = 142 known metabolites of Dendrobium species, was used to tentatively identify through dereplication the metabolites corresponding to each cluster of the heat map. After confirming or completing database proposals for all atom positions using an interpretation of 2D NMR data, 21 metabolites were identified in the D fimbriatum hydroethanolic extract. Cluster 1 corresponded to an intense group of 14 chemical shifts, which were assigned to dihydromelilotoside (

Hierarchical clustering analysis (HCA) correlation map of C-13 nuclear magnetic resonance (NMR) signals with the identified compound.
After the identification of the major constituents of the D fimbriatum extract obtained by the CARAMEL procedure, a targeted approach, using a classical HPLC purification procedure was employed to further characterize the minor and specialized chemical constituents.
Targeted HPLC Isolation
Several ultraviolet (UV) peaks at 280 nm were hypothesized to be stilbene derivatives due to maximum absorption at 260, 280, and 320 nm. 15 Stilbene derivatives such as phenanthrenes and bibenzyls are interesting chemical classes found mostly in Orchidaceae. They are recognized for several biological properties such as antiproliferative, antimicrobial, anti-inflammatory, and antioxidant activities. 16 Those metabolites were consequently targeted during the isolation procedure. For efficient separation and to obtain appropriate quantities for identification, the dry extract was fractionated by liquid–liquid extraction (LLE), leading to aqueous and ethyl acetate (EtOAc) fractions. Each fraction was separated by semipreparative liquid chromatography in reverse mode, targeting the isolation of the compounds responsible for the peaks at 280 nm in the EtOAc fraction and the main evaporative light scattering detector peak in the aqueous fraction. All compounds were finally identified based on their exact mass, fragmentation patterns, and NMR spectra.
From the aqueous fraction, fraction 19 yielded 12 mg of a furostanol saponin, identified as protodioscin
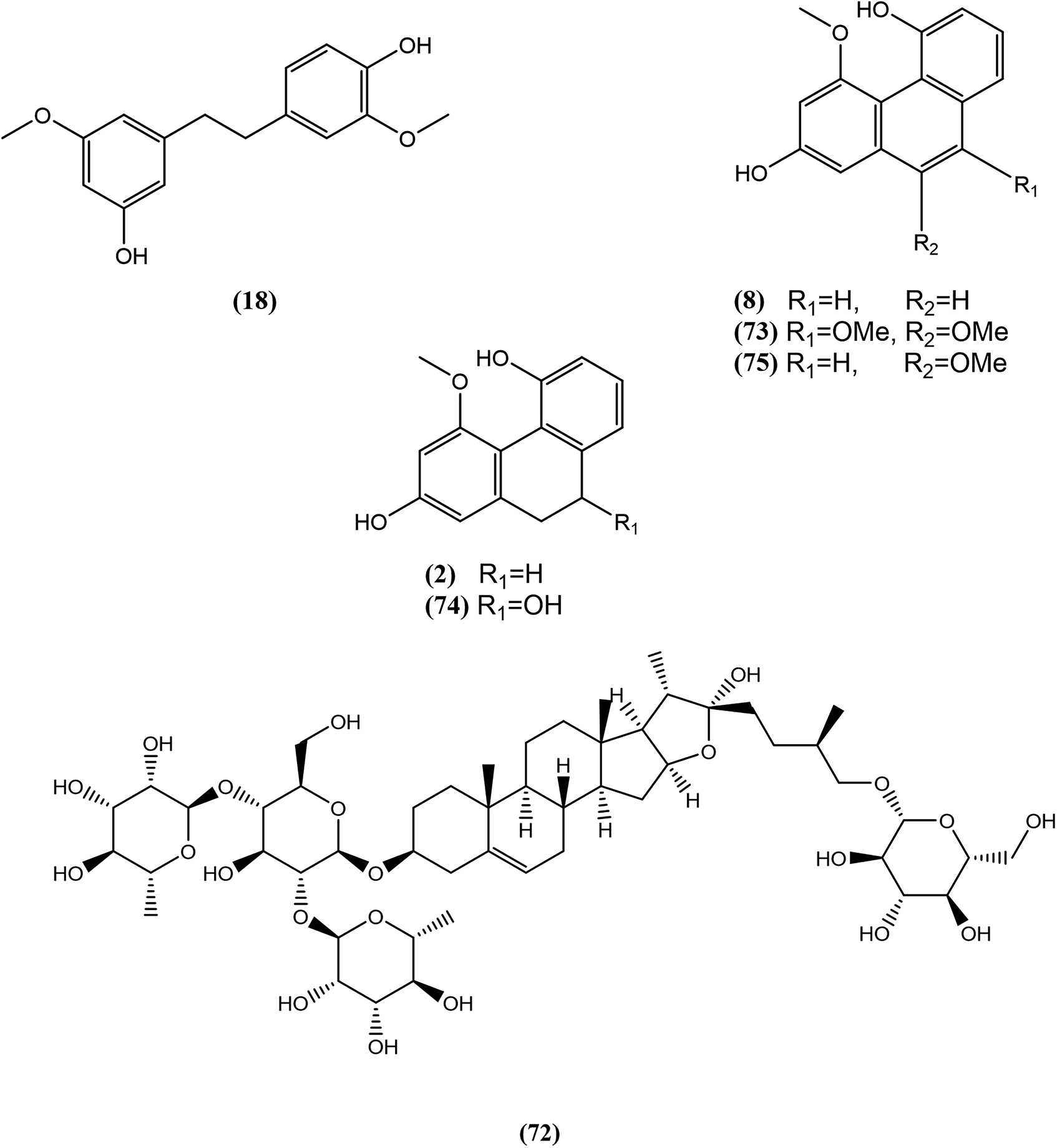
Structures of compounds isolated by semipreparative high-performance liquid chromatography (HPLC).
In addition, fraction F46 yielded a new phenanthrene derivative, 4, 10 dimethoxy-2,5-phenanthrenediol (3 mg), named plicatol D

NOE correlations of plicatol D
Accordingly, the structure of
Materials and Methods
Extraction
Air-dried whole plants of D fimbriatum (3 kg) were obtained from the Phrao Orchids Nursery green house in Chiang Mai, Thailand. The dried material was pulverized in a Wiley mill, providing 2.5 kg of a fine powder, and extracted 4 times with 50% ethanol under reflux. After filtration through paper, the filtrate was concentrated under vacuum and then lyophilized, leading to 750 g of a dry hydroethanolic extract.
CARAMEL Procedure
Extract fractionation by CPC
Two CPC experiments were performed successively. For the first CPC, 2073 mg of the extract was injected into a CPC column of 303 mL (FCPE300®, Rousselet Robatel Kromaton) containing 7 circular partition disks and engraved with a total of 231 partition twin cells (≈1 mL each twin cell). The liquid phases were pumped with a KNAUER Preparative 1800 V7115 pump. A 3-phase solvent system was prepared by mixing n-heptane, methyl-tert-butyl-ether, ACN, and water in the proportions 1/1/1/1 (vol/vol). The column was filled with the middle phase. After column equilibration with the upper phase at 20 mL/min at 1200 rpm, the dry extract was dissolved in 10 mL of lower phase + 5 mL of middle phase + 3 mL of upper phase and injected into the CPC column via a 20 mL sample loop. The upper phase (used as the mobile phase) was pumped in the ascending mode for 40 min at 20 mL/min. The column was then extruded by switching the mode selection valve for 20 min. Fractions of 20 mL were collected during elution and combined according to their thin layer chromatographic profiles. TLC was performed with a CAMAG® Automatic TLC Sampler 4 (ATS4), a CAMAG® Automatic Developing Chamber 2 (ADC2), and a CAMAG® TLC Visualizer 2. Fractions were deposited on precoated silica gel 60 F254 Merck plates, eluted with the migration solvent system ethyl acetate/toluene/acetic acid/formic acid (5/5/1/1, vol/vol), visualized under UV light at 254 nm and 366 nm, and revealed by spraying the dried plates with 50% H2SO4 and vanillin followed by heating. As a result, 4 fractions were obtained (Figure 2, from F01 to F04). All compounds retained inside the column were recovered by extrusion and dried under vacuum to obtain the second CPC injection (m = 1996 mg).
For the second CPC, 1996 mg was injected. The same solvent system was used, but the lower phase was used as the stationary phase and the middle phase was used as the mobile phase (ascending mode). The mobile phase was pumped in the ascending mode for 70 min and the column was extruded for 13 min. As a result, 9 subfractions were obtained (Figure 2, from F05 to F13). The mass distribution of all fractions obtained from CPC1 and CPC2 is also illustrated in Figure 2.
NMR analyses and identification of the major metabolites
All CPC fractions from F1 to F13 were dried under vacuum and an aliquot (up to 15 mg when possible) was dissolved in 700 µL of DMSO-d6 and analyzed by C-13 NMR at 298 K on a Bruker Avance III 600 spectrometer (H-1 at 600 MHz and C-13 at 151 MHz) equipped with a 5 mm TCI cryogenic probe (analytical platform PlAneT, University of Reims Champagne-Ardenne, Reims, France). Bruker TopSpin 4.0.5 software was used for NMR data acquisition and processing. A standard zgpg pulse sequence was used with an acquisition time of 0.9 s and a relaxation delay of 3 s. For each sample, 512 scans were co-added to obtain a satisfactory signal-to-noise ratio. The spectral width was 240 ppm and the receiver gain set to the highest value. Spectra were then manually phased, baseline corrected, and calibrated on the central resonance of DMSO-d6 (δ 39.8 ppm). The absolute intensities of all C-13 NMR signals detected in the spectra of all CPC fractions were collected by automatic peak picking, and each resulting peak list was exported as a text file. Then a binning step was performed using a script written in Python. Its principle was to divide the C-13 NMR spectral width into chemical shift windows of 0.3 ppm and to associate the absolute intensity of each peak to the corresponding bin. The resulting table was submitted to HCA using PermutMatrix 1.9.3 software (LIRMM) for data visualization. In parallel, a literature survey was performed to obtain the structures of a maximum of metabolites already reported for D fimbriatum (n ≈ 142). The C-13 NMR chemical shifts of these metabolites were predicted (NMR Workbook Suite 2012, ACD/Labs) and stored in an in-house database already comprising ≈ 6200 entries of natural products. The chemical shift clusters obtained by HCA were submitted to this database to initiate the identification process. Following this direct dereplication step, 2D NMR analyses (HSQC, HMBC, and COSY) were also interpreted to validate or further complete the structural elucidation of metabolites proposed by the database.
Targeted Semipreparative HPLC Procedure
Extract fractionation by LLE
The dry hydroethanolic extract (30 g) was resolubilized in water to obtain a 30 mg/mL solution and fractionated by LLE with ethyl acetate (EtOAc) (1/1, vol/vol). Three cycles of LLE with the organic solvent were performed. The aqueous and EtOAc phases were evaporated, leading to aqueous and EtOAC fractions, respectively.
Isolation of minor constituents by liquid chromatography
Semipreparative HPLC separation was performed using a Gilson 322 pump equipped with a UV/VIS-151 detector, a FC-204 Gilson fraction collector, and a C18 Axia column, 5 µm, 21.2 × 100 mm (Phenomenex). The mobile phase was comprised of ACN and water with 0.05% formic acid; the flow rate was set at 25 mL/min.
The EtOAc fraction (400 mg in total) was fractionated into 60 fractions using the semipreparative reverse-phase HPLC system. The gradient conditions were 5% ACN during 2 min, from 5% to 25% in 10 min, from 25% to 35% in 35 min, and from 35% to 100% in 5 min. The UV detection was set at 280 nm. This operation was repeated 10 times (40 mg injected each time). Similar numerical fractions were combined, and further purification was performed using different isocratic conditions with an Interchim PuriFlash column 5 μm, 10 × 250 mm, (ref PF5C18AQ, Interchim), with a flow rate of 6 mL/min.
The aqueous fraction (800 mg injected in total) was fractionated using the same equipment with the following gradient conditions: 5% ACN from 20% ACN during 22 min, from 22% to 55% in 12 min, and from 55% to 70% in 40 min. This operation was repeated 10 times (80 mg injected each time).
HRMS and NMR analyses of purified metabolites
High-resolution mass spectrometry (HRMS) data were obtained on a hybrid quadrupole time-of-flight mass spectrometer (microTOf Q II, Bruker) equipped with an electrospray interface and coupled with an Acquity UPLC system (Waters). The column was an Acquity UPLC BEH C18 1.7 µm, 2.1 × 100 mm.
A Bruker Avance Neo 600 MHz NMR spectrometer equipped with a QCI 5 mm cryoprobe and a SampleJet automated sample changer (Bruker BioSpin) were employed for 1D and 2D-NMR spectroscopy. Chemical shifts were reported in parts per million (δ) using the residual signal of the CDCl3 signal (δH 7.26; δC 77.2) as internal standards for H-1 and C-13 NMR, respectively, and coupling constants (J) were reported in Hz.
Retention times, mass fragmentation spectra, and H-1 and HSQC spectra of compounds
Plicatol D (4, 10 dimethoxy-2,5-phenanthrenediol): yellowish powder, UV (CHCl3) λmax nm (log ε): 256 (3.99), 279 (3.94), 323 (3.52); HRMS: 271.0966 [M + H]+ (C16H15O4) Δ1.5 ppm ; Table S2, 1H NMR (CDCl3, 600 MHz) δ 9.40 (1H, s, OH-5), 7.51 (1H, d, J = 2.5 Hz, H-1), 7.42 (1H, t, J = 7.7 Hz, H-7), 7.31 (1H, dd, J = 7.7, 1.4 Hz, H-8), 7.07 (1H, dd, J = 7.7, 1.4 Hz, H-6), 6.92 (1H, s, H-9), 6.89 (1H, d, J = 2.5 Hz, H-3), 4.07 (3H, s, 4-OMe), 4.04 (3H, s, 10-OMe); 13C NMR (CDCl3, 151 MHz) δ 55.7 (10-OMe), 58.7 (4-OMe), 101.7 (C-1), 102.3 (C-3), 104.6 (C-9), 114.6 (C-6), 115.2 (C-4b), 115.7 (C-4a), 119.5 (C-8), 127.5 (C-7), 130.7 (C-14), 134.7 (C-8a), 151.8 (C-10), 154.1 (C-5), 154.6 (C-2), and 155.6 (C-4). NMR spectra available in Figures S2 to S7.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221074526 - Supplemental material for Extensive Phytochemical Assessment of Dendrobium fimbriatum Hook (Orchidaceae)
Supplemental material, sj-docx-1-npx-10.1177_1934578X221074526 for Extensive Phytochemical Assessment of Dendrobium fimbriatum Hook (Orchidaceae) by Quentin Favre-Godal, Jane Hubert, Alexis Kotland, Delphine Garnier, Camille Beaugendre, Lorène Gourguillon, Aurelie Urbain, Sonia Lordel-Madeleine and Patrick Choisy in Natural Product Communications
Footnotes
Acknowledgments
This work was supported by Guerlain & LVMH Recherche to contribute to the understanding and preservation of orchids. The authors are thankful to Léa Mazina for her logistic support, and to Laurence Marcourt from the Institute of Pharmaceutical Sciences of Western Switzerland (ISPO) of the University of Geneva (Prof. J-L. Wolfender) for her help in NMR experiments.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.