
Abstract
Introduction
A novel coronavirus disease that caused an acute infectious pneumonia outbreak at the end of 2019 was named COVID-19 by WHO. As of May 28, 2021, millions of individuals have died from COVID-19 worldwide, and many countries are still fighting against it. The new type of COVID-19, belonging to the category of epidemic diseases in TCM, causes lesions in the lung. 1 The concept of TCM holds that COVID-19 is mainly caused by the invasion of an external pathogen (SARS-COV-2) and deficiency of a healthy qi (decline of human immunity). 2 Many TCMs have been reported to have good therapeutic effects on patients with COVID-19 at different stages, accumulating rich evidence for the use of these TCMs. The clinical manifestations of patients showed that COVID-19-related changes had obvious periodicity. In the COVID-19 clinical protocol eighth edition published by the People's Republic of China, the clinical symptoms of COVID-19 were divided into five categories: mild, moderate, severe, critical and convalescent. Han-Shi-Yu-Fei (HSYF), Han-Shi-Zu-Fei (HSZF), and Yi-Du-Bi-Fei (YDBF) formulas were recommended to treat mild, moderate, and severe symptoms in this clinical protocol, respectively. 3 Because the active components and mechanism of these TCMs in the treatment of COVID-19 are still uncertain, their promotions and applications are hindered.
Network pharmacology is a combination of systems biology, multidirectional pharmacology, and computer analysis technology. Molecular docking is the main tool used to study the interaction between drugs and proteins and is widely used in the research and development of new drugs.4,5 With the rapid development of these tools, the active components and mechanisms of TCMs have gradually been explored. Therefore, in this study, network pharmacology and molecular docking were used to explore the active components and mechanisms of these three prescriptions (HSYF, HSZF, and YDBF) to expand their promotion and application. Through the construction of “C-T-P” and “PPI” networks, the active components and potential targets were explored. Molecular docking proved the binding energies between active components and ACE2 were lower than the threshold, suggesting that these components could inhibit the binding of ACE2 and SARS-COV-2 3CL to protect against COVID-19. The workflow of this study is shown in Figure 1.

Flow chart of this study.
Materials and Methods
Collecting Potential Components
The objective was to determine the overlapping TCMs and collect their potential components in HSYF, HSZF, and YDBF prescriptions. After determining the TCMs in each prescription, Venn 2.1.0 database was used to analyze and obtain their intersection. TCMSP (http://lsp.nwu.edu.cn/tcmsp.php) was used to collect all their components. Based on the guidance of TCMSP, the active components were screened using the index of OB≥30% and DL≥0.186,7 to ensure that all components that could be developed into drugs were not omitted to the greatest extent. Active components’ common names and PubChem CID were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) to establish a chemical composition database of these TCMs. Finally, the “P-T-C” network was established using Cytoscape 3.7.1 software to analyze the relationship between them.
Collecting Targets of Potential Components
SwissTargetPrediction database (http://www.swisstargetpredict ion.ch/index.php) was used for collecting the potential targets related to the active components. Using the Retrieve/ID mapping tool and setting the species as “human,” the Gene Official Symbol formats of the candidate targets were obtained. Subsequently, Gene Cards (https://www.genecards.org/) was used to identify the targets related to COVID-19 by searching the keywords “COVID-19” and “Novel coronavirus pneumonia”. Finally, the overlapping targets of COVID-19 and active components were analyzed using the Venn 2.1.0 database.
Construction of “Protein–Protein” Network
The targets of TCMs in the treatment of COVID-19 were analyzed by STRING database (https://string-db.org/), and their corresponding species were set to “Homo sapiens”. Following this, the interaction between targets was analyzed, and the “PPI” network was downloaded. To ensure the integrity and accuracy of the whole “PPI” network analysis process, a TSV format file was imported into the Cytoscape 3.7.1 software, and their degrees of interaction were analyzed through the plug-in CytoHubba. The candidate targets for which the degree of node exceeded the average were selected.
Gene Ontology and KEGG Pathway Analysis
The DAVID 6.8.1 database (https://david.ncifcrf.gol/) is a commonly used tool for analyzing the biological functions and pathways of proteins. Selecting the identifier as “GENE OFFICIAL SYMBOL” and species as “Homo sapiens,” the candidate targets were imported into DAVID 6.8.1 for GO entries and KEGG pathway annotations. With P < 0.05, the top ten biological process (BP), cellular component (CC), and molecular function (MF) entries were plotted as circos using the Omicshare database (http://www.omicshare.com). Further, the top ten KEGG pathways were plotted as bubble plots using the R software package.
Construction of the “Components-Targets-Pathways” Network
Based on the active components, candidate targets, and top ten KEGG pathways, the “C-T-P” network was constructed with Cytoscape 3.7.1 software. In this network, nodes represent the potential components, targets, and pathways, whereas edges represent the interactions between these biological nodes. The degrees of these nodes and edges are usually used to represent the number of connections between components and targets in the network core architecture. 8 Therefore, the top five components were selected as molecular docking ligands according to the ranking of degree. Further, the top five targets related to components and pathways were screened.
Correlation Analysis of Candidate Targets and ACE2
ACE2, also known as Aceh, is a receptor for the virus to enter the host cell, and its biological process is to interact with the host virus. A large number of studies have shown that its encoding protein is the functional receptor of SARS-CoV and SARS-CoV-2 S glycoprotein. Because ACE2 shows stable binding with SARS-CoV-2 and SARS-CoV, it is considered to have good molecular recognition, binding and dynamic process functions. The analysis of the correlation between the above-mentioned targets and ACE2 can confirm the role of these targets in COVID-19 and judge the accuracy of previous results. Therefore, STRING was used to analyze the correlation between them in the form of a “PPI” network. After obtaining the targets with a strong correlation with ACE2, R language was used to analyze the correlation between them and the top five targets in the “C-T-P” network in the form of a heat map. Based on the above-mentioned results, the core targets were obtained.
Molecular Docking
The reported 3D structures of SARS-COV-2 3CL and ACE2 were downloaded from the RSCBPDB (https://www.rcsb.org/) database. The 3D structures of the active components were downloaded from the PubChem (https://pubchem.ncbi.nlm.nih.gov/) database. The 3D structures were drawn and generated using Chem draw software. Following this, according to the MM2 molecular mechanics algorithm, the commands of calculations > MM2 > minimize energy were executed successively to minimize the energies of all components. The iterative information and structural parameters of each component were outputted in turn. The ligands and non-protein molecules (such as water molecules) in these two targets were removed using PyMol software. After adding H atoms to these components and proteins, the AutoDockTools-1.5.6 software was used to calculate the docking energies. The docking results were analyzed to show the functional groups and amino acid residues at the binding sites of components and targets by the PyMol software. Finally, the binding sites of ACE2 and 3CL were searched from literature and the PDB database, and these were compared with the molecular docking results.
Results
Overlapping Traditional Chinese Medicines and Potential Components of These Three Prescriptions
Based on the composition of the three prescriptions (HSYF, HSZF, and YDBF), a total of five overlapping TCMs were obtained. They were Ephedrae Herba, Pogostemonis Herba, Atractylodis Rhizoma, Magnoliae Officinalis Cortex, and Tsaoko Fructus. According to the concept of TCM, they were all important TCMs in these three prescriptions. On referring to the TCMSP database, 40 chemical components were obtained by setting OB≥30% and DL≥0.18 as the threshold. The relationship between the prescriptions, TCMs, and potential components is shown in Figure 2, and detailed information on the components is shown in Table 1.

The network of “P-T-C”. Purple nodes represent prescriptions, pink nodes represent overlapping TCMs, and green nodes represent components.
Detailed Information of Potential Components.

Potential Targets of Components
After removing repeated targets, 741 related targets of the potential components were obtained from the SwissTargetPrediction database. Pie graphs of all components are shown in Supplementary Figs 1 and 2. Meanwhile, 659 targets of COVID-19 were screened for “novel coronavirus pneumonia” and “COVID-19” according to the score in the Gene Cards database. Using the Venn 2.1.0 database, 92 overlapping targets were obtained, as shown in Figure 3A. The specific information of them is shown in Table 2.
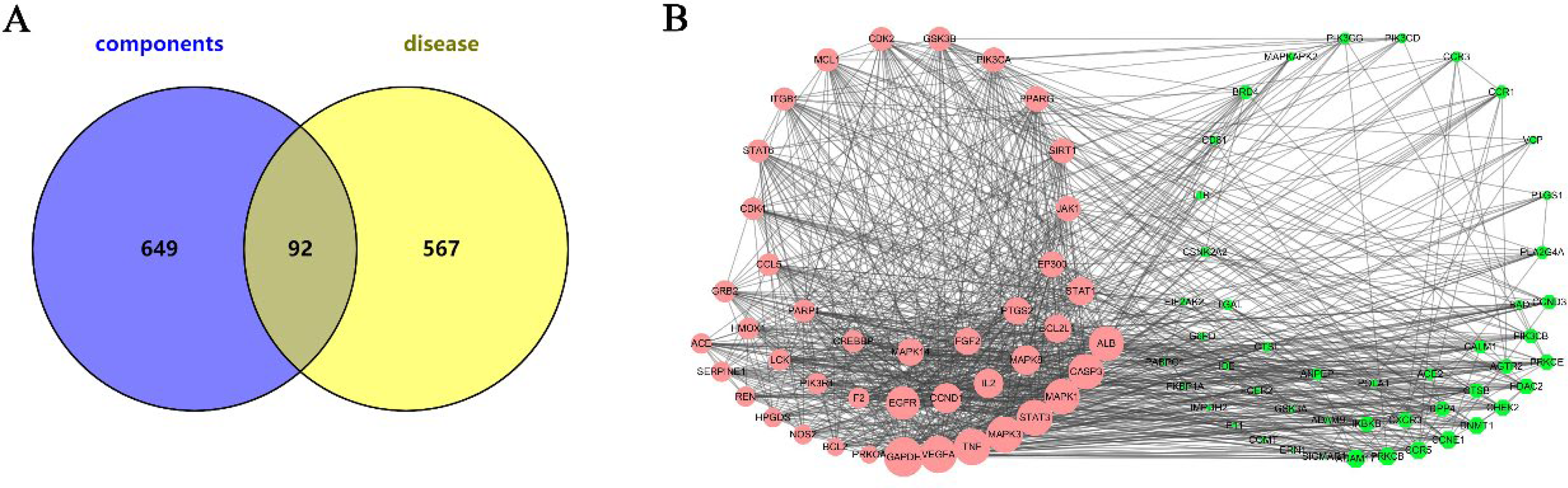
Venn diagram and “PPI” network. (A) Venn diagram of components and COVID-19, (B) “PPI” network of candidate targets.
Potential Active Gene Targets.

Construction of the “Protein–Protein” Network
When the targets were visually analyzed by Cytoscape 3.7.1, the calculation rules of this software were used to analyze the degrees of nodes. The degree is the most direct measure of node centrality in network analysis. The greater the degree of a node, the higher its degree of centrality, and the more important the node is in the network. Accordingly, a total of 47 hub candidate genes were obtained by analyzing 92 overlapping targets with the plug-in CytoHubba in Cytoscape 3.7.1; Figure 3B shows the details. The depth of node color and the node size were positively correlated with their degrees.
Biological Function Analysis
Analyzing the biological functions of these 47 candidate genes and the pathways they participate in is the key to understanding the relationship between them and COVID-19. Through GO function enrichment analysis, 393 GO entries were obtained with the threshold of P < 0.05. Among them, there were 211 BP entries, 30 CC entries, and 47 MF entries. The first 10 entries of these were drawn into a circos, as shown in Figure 4A. BP and CC enrichment analysis in GO showed that they mainly regulated the synthesis of the cytoplasm and nucleus, and affected cell proliferation, apoptosis, and RNA synthesis. The lung injury induced by COVID-19 was closely associated with the inhibition of cell proliferation and the aggravation of apoptosis. The MF entries showed that they could regulate protein kinase, protein binding, and kinase activities, which played important roles in the above-mentioned processes. These results confirmed that these targets could promote the occurrence and development of COVID-19.
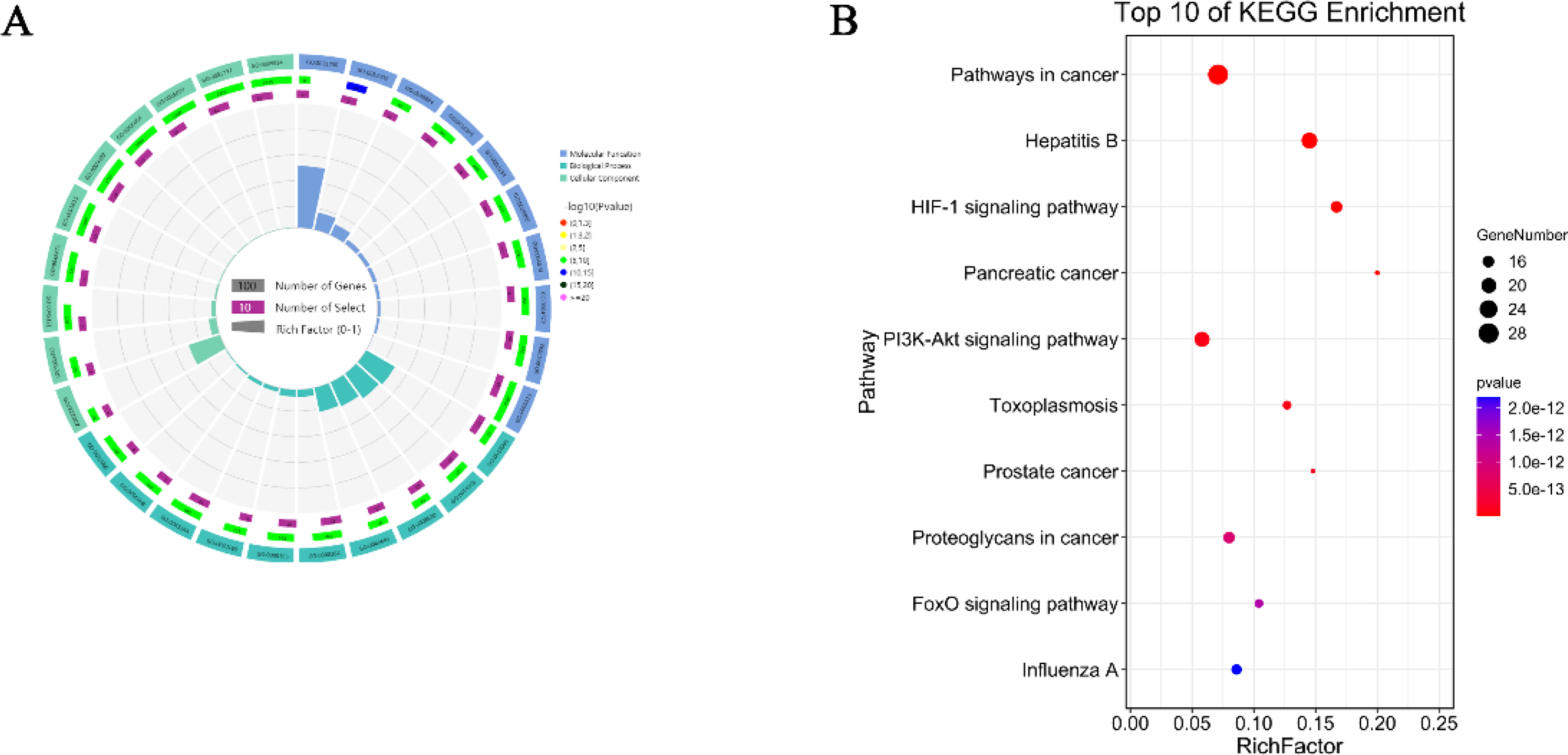
Enrichment analysis of GO and KEGG. (A) Circos of top ten GO BP, GO MF, and GO CC entries; (B) bubble chart of the top ten KEGG pathways.
The results of KEGG pathway analysis showed that there were 105 pathways (P < 0.05), including pathways in cancer, hepatitis B, HIF-1 signaling pathway, and others. It was not difficult to find that they were mainly involved in such important pathological processes as virus infection, immune cell differentiation, and signal transduction pathways. The importance of these processes in COVID-19 is self-evident, and many studies have confirmed the correlation between these pathways and the corresponding symptoms of COVID-19. A bubble plot of the 10 most significant KEGG pathways is shown in Figure 4B.
Construction of “Components-Targets-Pathways” Network
Using Cytoscape 3.7.1, 40 potential components, 47 candidate targets, and the top 10 KEGG pathways were collected to construct a “C-T-P” network. As shown in Figure 5, purple nodes represent KEGG pathways, green nodes represent components, and rose-red nodes represent candidate targets. The first five components with higher degrees were selected as the important ligands in the subsequent analysis depending on the condition that the degree was more than twice that of the median. The top five components were diisooctyl phthalate, Q27134551, acanthoside B, neohesperidin, and irisolidone. It is generally believed that the importance of the role of a target between components and pathways is positively associated with its degree. Therefore, according to the ranking of the targets’ degrees, the top five targets were selected. These were EGFR, PTGS2, CDK2, GSK3B and PIK3R1.
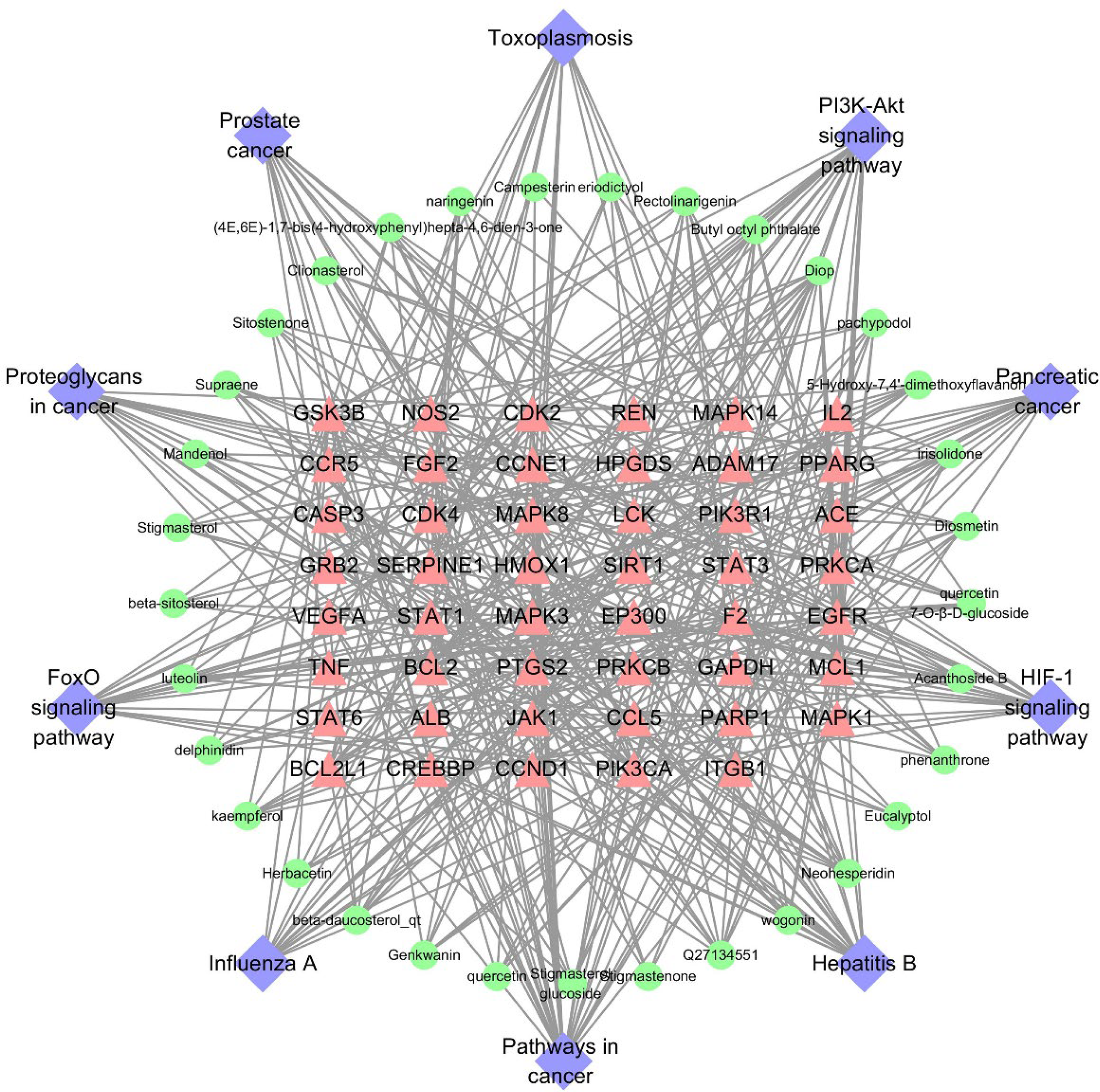
The network of “C-T-P”. Purple nodes represent KEGG pathways, pink nodes represent candidate targets, and green nodes represent components.
However, on searching related literature, diisooctyl phthalate was found to be an artifact rather than a real component of these TCMs. After removing diisooctyl phthalate and its targets from the “C-T-P” network, the final targets were set and the degree ranking of each target did not change. Therefore, it did not affect the mining results of core targets and was removed from the molecular docking research.
Selection of Core Targets
The correlation analysis between candidate targets and ACE2 showed that MAPK3, ALB, CDK4, REN, ITGB1, and ADAM17 were more closely associated with ACE2 than others in the “PPI” network according to their degrees. To identify further the core targets, R software was used to analyze the correlation between these six targets and the top five targets in the “C-T-P” network. The heat map results showed that there was a strong correlation between all of them. Based on the above-mentioned results, ALB, EGFR, MAPK3, ITGB1, and PTGS2 were considered to be the connection hubs of ACE2, active components, and KEGG pathways (Figure 6).

Screening of core targets. (A) “PPI” network of ACE2 and candidate targets, (B) “PPI” network of candidate targets, and (C) Heat map of important targets.
Molecular Docking and Analysis
According to the official tutorial of auto dock, the components with high degrees were docked with SARS-COV-2 3CL and ACE2. Docking energies and related docking residue sites are shown in Table 3 and Figures 7 and 8, respectively. It is generally believed that the lower the docking energy between components and proteins, the stronger their bond. The results showed that Q27134551, acanthoside B, neohesperidin, and irisolidone all had stable binding abilities with ACE2 and SARS-COV-2 3CL. Among them, Q27134551 and irisolidone had the lowest docking energy with ACE2 and SARS-COV-2 3CL, indicating that they could stably bind to these two proteins. According to Table 3, the binding amino acid residues between these components and ACE2 was LYS-94. Therefore, LYS-94 was an important amino acid site for the development of ACE2 inhibitors.

Molecular docking diagrams of ACE2 with active components. (A) Acanthoside B, (B) Irisolidone, (C) Neohesperidin, and (D) Q27134551.

Molecular docking diagrams of SARS-COV-2 3CL with active components. (A) Acanthoside B, (B) Irisolidone, (C) Neohesperidin, and (D) Q27134551.
Docking Energies (△Gb) and Bonds Between Components and Proteins.

Analyzing the binding residue sites of ACE2 and SARS-COV-2 3CL and comparing whether these components have the ability to prevent the binding of these two proteins are the key to judge whether these components could play important roles in the treatment of COVID-19. From the PDB database, 9 the binding hotspots of SARS-CoV RBM/hACE2 interface were hotspot-31 and hotspot-353, and the two virus-binding hotspots have become more stabilized in the SARS-CoV-2 RBM/hACE2 interface. The hotspot-31 is composed of LYS-31 and GLU-35, and the hotspot-353 is composed of LYS-353 and ASP-38. On comparing the above-mentioned results, it was not hard to determine that the sequence numbers of the docking residue sites in the docking results were inconsistent with the above-mentioned findings. However, this result did not negate the accuracy of the previous molecular docking results, but suggested that these binding residue amino sites may be new sites that have not been found yet. However, the accuracy of this conclusion needs to be demonstrated by further in vivo and in vitro experiments.
Discussion
In China, HSYS, HSZF, and YDBF have been used to treat patients with COVID-19 with mild, moderate, and severe symptoms, respectively. Because the active components and mechanism of these prescriptions are not clear, their clinical applications are hindered. In this study, these problems were analyzed via network pharmacology and molecular docking.
A comprehensive analysis of the TCMs in these three prescriptions revealed that they have five overlapping TCMs. Modern pharmacological research showed that Magnoliae Officinalis Cortex and Atractylodis Rhizoma have anti-lung injury, cough-relieving, sputum-removing, anti-inflammatory, and antiviral effects.10,11 Ephedrae Herba and Pogostemonis Herba could act against asthmatic and viral effects. 12 Furthermore, Tsaoko Fructus has anti-infection and anti-inflammatory effects in vivo.13,14 COVID-19 causes lung injury and aggravates inflammatory reaction and virus infection after attacking the human body. The results of these studies confirmed that all the five TCMs played important roles in the treatment of COVID-19.
Based on the guidance of TCMSP, the active components were screened using the index of OB≥30% and DL≥0.18. Their potential targets in the treatment of COVID-19 were screened via several network pharmacology databases. Following this, the DAVID 6.8.1 database was used to analyze the functions and pathways of these targets to explain their roles in COVID-19. GO enrichment analysis showed that they mainly acted on the nucleus and cytoplasm, and affected protein binding, protein phosphatase binding, and kinase activities. Thus, these targets played very important roles in regulating cell proliferation, apoptosis, and RNA polymerase II promoter transcription. A number of studies have shown that acute and chronic lung injury can cause cellular apoptosis in the lungs, and slow down the proliferation of normal cells, resulting in lung dysfunction. The KEGG pathways results showed that the potential targets were mainly involved in viral infection, immune cell differentiation, signal transduction, and other important pathological processes. COVID-19 has been confirmed to be caused by a virus infection. Therefore, reducing or preventing virus infection is the key to protect against it. Immune ability plays an important role in the process of the body's response to an attack by SARS-CoV-2. When the signal transduction mechanism is activated, it will aggravate the production of inflammation factors and viral infection, thus causing more serious damage to the host. Therefore, the key to COVID-19 treatment is to prevent viral infection, enhance the body's immunity and block the further transmission of related factors.
The results of the “C-T-P” network showed that diisooctyl phthalate, Q27134551, acanthoside B, neohesperidin and irisolidone were the top five potential components. However, diisooctyl phthalate was found to be an artifact rather than a real component of these TCMs, according to several related literature reports. Further, the final target set and the degree ranking of each target did not change when diisooctyl phthalate and its targets were removed from the “C-T-P” network. Therefore, it was not considered to affect the mining results of core targets and was removed from the molecular docking research. On analyzing the degrees of the nodes in the “C-T-P” and “PPI” networks, we found that ALB, EGFR, ITGB1, MAPK3, and PTGS2 were connected with more targets than the others. Therefore, they were the hubs of these targets, ACE2, active components, and pathways.
Epigenetics is a relatively new scientific field. 15 It regulates the expression of host genes through a series of reversible epigenetic modifications such as histone methylation and acetylation, and DNA/RNA methylation.16,17 After infection, the virus regulates the epigenetic landscape of host cells to resist cellular signals, host immunity, and antiviral ability, thereby enhancing its replication and infection efficiencies. Studies have shown that the methylation of ACE2 and post-translational histone modifications may lead to undifferentiated virus infection in the host.18,19 Methylation of ALB and ITGB1 can aggravate the risk of tumors induced by HBV. The expression levels of hubs are usually associated with liver dysfunction and inflammatory state, 20 which was consistent with the clinical symptoms of patients with COVID-19. Venkataraman 21 established a mouse model of SARS-CoV pathogenesis to explore the role of key targets. The results showed that EGFR played a key role in the host response to SARS-CoV. In EGFR (DSK5) mice, a SARS-CoV infection could aggravate lung disease, and EGFR ligands amphiregulin and heparin-binding EGF-like growth factor (HB-EGF) were upregulated during infection. These findings indicated that the wound repair pathway, controlled by the epidermal growth factor receptor (EGFR), was crucial for the recovery of SARS-CoV-induced tissue injury. The high homology between SARS-CoV and SARS-CoV-2 3CL suggested that EGFR may also play an important role in COVID-19, and its expression is regulated by promoter DNA methylation. Research has shown 22 that genetic deletion of the CCL2 receptor in mice with β1 integrin-deficient type 2 AECs inhibited the aggregation of monocyte-derived macrophages, leading to accelerated inflammation and severe premature emphysematous destruction. A selective inhibitor of PTGS2, also known as COX-2, was shown to have potential efficacy in high-risk patients with COVID-19, and PTGS2 methylation could accelerate the replication of HPV, HBV, and other viruses. 23 This further indicated that PTGS2 played an important role in the treatment of COVID-19 and viral disease. MAPK3 has been confirmed to be associated with hepatitis C virus-infected hosts. At the same time, it has also been confirmed to be significantly co-expressed with ACE2, which has been recorded in Swiss web. These studies confirmed that these targets played important roles in the occurrence and development of COVID-19, and also confirmed the accuracy of the results of network pharmacology.
It is well-known that the main aim of the treatment of COVID-19 is to block the virus from biotransformation in the body. Accordingly, it is important to find drugs targeting ACE2 and SARS-COV-2 3CL. The docking results of SARS-COV-2 3CL and ACE2 with the active components of these three prescriptions showed that the top four components in the network had strong binding abilities to ACE2 and SARS-COV-2 3CL. It is worth noting that Q27134551 and irisolidone have stable binding abilities with them. Q27134551 formed three hydrogen bonds with amino acid residues LYS94, TRP566, and GLN98. Furthermore, irisolidone formed two hydrogen bonds with LYS94 and ASN210, thus forming a stable complex with ACE2. Moreover, Q27134551 formed one hydrogen bond with LYS100, and irisolidone formed three hydrogen bonds with HIS163, THR26, and CYS145, thus forming stable complexes with SARS-COV-2 3CL. In comparison with the two hotspots in PDB and the literature, this study indicated that LYS94 was a key residue during docking with ACE2. Kim showed that the fermented extract of Schisandra chinensis had strong anti-inflammatory activity, and contained acanthoside B. 24 The specific performance was the reduction of nitric oxide (NO) production, inhibition of iNOS, and COX-2, pro-inflammatory cytokines (IL-6), and tumor necrosis factor (TNF-2). At the same time, another study showed that neohesperidin antagonized TGF-β1/Smad3 signaling and played a role in idiopathic pulmonary fibrosis. 25 It could not only inhibit the TGF-β1-induced injury in alveolar epithelial cells, but also decrease the TGF-β1-induced myofibroblast differentiation, extracellular matrix production, and fibroblast migration. Irisolidone inhibited the production of TNF-α and IL-1β and inflammatory mediators NO and PGE2 via the NF-κB pathway, which indicated that it could inhibit inflammatory reactions. 26 These studies indicated that the top five components had potential activities in the treatment of COVID-19. At present, there are few studies on diisooctyl phthalate and Q27134551, and their active mechanism needs further investigation.
Conclusion
Taken together, this study revealed four active components, three core targets, one important amino acid site and the potential mechanism of three clinical prescriptions against COVID-19 via network pharmacology and molecular docking. These findings provide a strong theoretical basis for the anti-COVID-19 effect of these prescriptions and lay a solid foundation for the promotion of their application in the treatment of COVID-19.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X211047702 - Supplemental material for Analysis of the Active Components and Mechanism of Three Prescriptions in the Treatment of COVID-19 Via Network Pharmacology and Molecular Docking
Supplemental material, sj-docx-1-npx-10.1177_1934578X211047702 for Analysis of the Active Components and Mechanism of Three Prescriptions in the Treatment of COVID-19 Via Network Pharmacology and Molecular Docking by Fei Wang, Jia-Hui Chen, Bo Liu and Ting Zhang in Natural Product Communications
Footnotes
Acknowledgments
We sincerely thank all colleagues who advised on the process of writing the paper.
Author Contributions
Fei Wang and Jia-Hui Chen completed the data mining of the article and the writing of the first draft, Bo Liu and Ting Zhangcompleted the polishing of the article and the proofreading of the data.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Hubei Province (grant number, 2020CFB535).
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.