
Abstract
Spiroacetals constitute the central structural core element of numerous natural products and are mostly represented as bicyclic or tricyclic domains. Typical natural products with tricyclic spiroacetals are (+)-ranuncoside
A multitude of natural products of insect, marine, bacterial, or plant origin is characterized by spiroacetal domains as a central structural core element.
1
-3
Generally, these central domains are represented as bicyclic and tricyclic spiroacetal ring systems which possess complex stereochemistry and specific biological activities. While investigating natural bioactive phytoagents, Tscheche et al
4
described that the 2 toxic plants Ranunculus repens and Helleborus foetidus contain glycosides with lactone forming aglycon which displays considerable antibiotic properties. They extracted a novel crystalline compound with positive rotation from a pool of active substances and named it ranuncoside due to its origin from Ranunculaceae species. The same group elucidated its structure by peracetylation and subsequent analysis of its 90 MHz 1H NMR spectrum. (+)-Ranuncoside

The plant glycoside (+)-ranuncoside 1, the marine algal toxins (+)-okadaic acid 2, and the (+)-dinophysistoxins-1 and 2 (structures 3 and 4) are characterized by a tricyclic spiroacetal domain (highlighted in red).
The biosynthesis of
Confusingly, the name ranuncoside was later also used for 2 completely different structures: (1) a pentacyclic triterpenoid glycoside from Hydrocotyle ranuncoloides (Apiaceae) 9 and (2) an olefinic glycoside from Ranunculus muricatus (Ranunculaceae) with unsolved stereochemistry in the whole hexose moiety. 10
(+)-Okadaic acid
It is interesting to note that terrestrial plants and algae produce different tricyclic spiro ring systems. While (+)-ranuncoside

Structures and stereochemistries of the tricyclic spiroacetal domain of (+)-ranuncoside 1 (structure 5), of the segment B of (+)-okadaic acid 2 and the (+)-dinophysistoxins 3 and 4 (structure 6). The corresponding structural analogous domains, 7 and (ent)
Due to the remarkable bioactivity of numerous natural compounds with spiroacetal domains,
18
we developed the synthesis of novel bicyclic spiroacetal motifs by highly stereoselective spiroacetalization of pyranodioxanes.
19,20
Here, we adapted this methodology to linear fused pyran-dioxane-cyclohexane tricycles in order to obtain tricyclic spiroacetal domains, since we considered it to be the most logical and feasible approach. Of the derivatives we aimed to synthesize by this methodology, particularly those with the spiro domain
Results and Discussion
The chemical elaboration of derivatives with spiro furan-dioxane-cyclohexane ring system

Hydroxylation of cneorine C (8) with osmium tetroxid to the cis-diol 9 followed by sodium periodate cleavage to the bis-half acetal
Our stereoselective synthesis of derivatives of the spiroacetal
Synthesis of Pyran-dioxane-cyclohexane Tricycles 12 , 17 , and 25
In 2004, we published the synthesis of pyran-dioxane bicycles through bisacetalic annulation of 2-ketosugars to glycol.
19
In a directly connected paper, we described the glycosylation of (R,R)-configured and (S,S)-configured cyclohexane-1,2-diols with ulosyl bromide
Here, we describe an established 50 g synthesis of

α-cis- or β-cis-Addition of (R,R)-cyclohexanediol on ulosylbromide 11 gave the pyran-dioxanecyclohexane tricycles 12 and 17. Thus, protonation of 12 (structure 13), ring contraction (structure 14), water addition (structure 15), and final benzoylation yielded 16. Based on 17
22
as starting material, the reaction sequence was identical and differed only by the primary epimerization of

All-chair conformations of (a) the spiroacetal derivatives 16, 24, and 29, indication the anomeric effects by blue arrows and (b) of the starting material 12, illustrating the operative nuclear Overhauser effect proven by 1H-nuclear magnetic resonance spectroscopy.
Glycosylation of (R,R)-Cyclohexanediol With Ulosylbromide 11: Reaction Conditions, Ratio of Product 12 and 17, as well as Overall Yield.

Abbreviations: Ag2CO3, silver carbonate; AgOTf, silver triflate; CH2Cl2, dichloromethane; DMF, dimethylformamide; THF, tetrahydrofuran.
aDetermined by1H-nuclear magnetic resonance.
bOverall yield.
The isomeric pyran-dioxane-cyclohexane tricycle
In contrast to the 2 tricycles above, the isomer
Synthetic Access to the Spiroacetal Domain Derivative of (+)-Ranuncoside 1
Ring contraction of
Application of the same reaction conditions to the diastereomeric linear fused tricycle
In summary, both linear fused tricycles with (R,R)-cyclohexane portion, that is, compounds
Synthetic Access to the Spiroacetal Domain Derivative of (+)-Okadaic Acid 2 and the (+)-Dinophysistoxins-1 and 2 (3 and 4)
Reaction of the linear fused pyrane-dioxane-cyclohexane tricycle

Ring contraction of tricycle
The 2 synthesized spiroacetals
Structural Elucidation
Structure and all-chair conformation of the synthesized starting material, the pyran-dioxane-cyclohexane tricycle
The final products, the spiro furan-dioxane-cyclohexane tricycles
The furan chemical shifts and coupling constants, 3′-H, 4′-H, 5′-H, as well as of the spiro C-2 resonance of spiroacetals
Selected 1H NMR Data and 13C NMR Shifts of the Spiro Furan-Dioxane-Cyclohexane Tricycles 16 and 29.

Abbreviation: NMR, nuclear magnetic resonance.
aThe complete data are listed in the Experimental section.
Conclusions
The plant glycoside (+)-ranucoside
The 2 new spiroacetal domain derivatives of ranuncoside, and of okadaic acid and dinophysistoxin-1 and 2, the compounds
The structure of the new compounds, the starting material
Experimental
Thin-layer chromatography (TLC) was performed on POLYGRAM SILG/UV254 (Macherey Nagel & Co.). Preparative chromatographic separations were carried out on columns with Merck silica gel 60 (15-40 µm) and Merck precoated silica gel plates 60 F254, 20 × 20 cm, 0.25 mm. Melting points were determined on a Bock-Monoskop VS or on a Büchi SMP-20 and are uncorrected. Specific optical rotations were determined on a Perkin-Elmer Polarimeter 241 in 1 dm cuvettes at a wavelength of 589 nm. NMR spectra were measured on a Bruker WM 300 and DRX 500 spectrometer at 303 K using trimethylsilane as internal reference or by calibration with the shift of the solvent of the sample. 24 The abbreviation of the multiplicities of the shifts are indicated as s for singlet, d for doublet, t for triplet, q for quartet, sxt for sextet, oct for octet, m for multiplet, br for broad, and p for pseudo. Mass spectra were run on a MAT 311 mass spectrometer (Varian). Elemental analyses were performed on a Perkin Elmer 240 Elementar Analyser.
(+)-(2R,3R,4S,4aR,5aR,9aR,10aS)-3,4-Bis-Benzoyloxy-2-(Benzoyloxy)methyl-4a-Hydroxy-Decahydro-2H-Pyrano[2,3-b][1,4]benzodioxin (12)
Dimethylformamide without a silver salt promoter
A mixture of ulosylbromide
C33H32O10 (588.6): calcd. C 67.33, H 5.48; found C 67.14, H 5.53.
MS (FD, 20 mA): m/z = 588 (100%, M+).
1H NMR (500 MHz, CDCl3) δ 1.00-1.95 (m, 8H, 6-H2, 7-H2, 8-H2, 9-H2), 3.33 (ddd, 1H, 9a-H), 3.78 (ddd, 1H, 5a-H), 4.19 (br. s. 1H, 4a-OH), 4.36 (dd, 1 H, 2′-HA), 4.50 (ddd, 1H, 2H, in part overlapped with 2′-HB), 4.53 (dd, 1H, 2′-HB, in part overlapped with 2H), 4.79 (s, 1H, 10a-H), 5.84 (pt, 1H, 3H), 6.10 (d, 1H, 4H), 7.13-7.45 (m, 9H, aromatic meta- and para-H), 7.75-8.00 (m, 6H, aromatic ortho-H); J 2,2′A = 4.7, J 2,2′B = 3.0, J 2′gem = 12.5, J 2,3 = 10.0, J 3,4 = 9.7, J 5a,6ax = 10.6, J 5a,6eq = 4.2, J 5a,9a = 9.1, J 9a,9ax = 11.3, J 9a,9eq = 4.3 Hz.
NOE interaction 4H (6.10) ↔ 5a-H (3.78), 9a-H (3.33) ↔ 10a-H (4.79) ppm.
13C NMR (125.8 MHz, CDCl3) δ 24.05 (C-7 and C-8, overlapped), 29.72 (C-6 and C-9, overlapped), 63.32 (C-2′), 68.97 (C-3), 69.55 (C-4), 70.83 (C-2), 75.90 (C-5a), 79.88 (C-9a), 94.24 (C-4a), 98.69 (C-10a), 128.0-130.5 (3 C 6H5CO), 165.39, 165.97, 166.45 (3 C6H5 CO).
Silver triflate in tetrahydrofuran
A solution of (1R,2R)-trans-1,2-cycohexanediol (116 mg, 1.00 mmol) in anhydrous tetrahydrofuran (10 mL) was cooled down to 0 °C. The solutions of ulosylbromide
Silver triflate in dichloromethane
A mixture of (1R,2R)-trans-1,2-cycohexanediol (116 mg, 1.00 mmol), ulosylbromide
(+)-(2R,3R,4S,4aS,5aR,9aR,10aR)-3,4-Bis-Benzoyloxy-2-(Benzoyloxy)methyl-4a-Hydroxy-Decahydro-2H-Pyrano[2,3-b][1,4]benzodioxin (17)
Silver carbonate in dichloromethane
For experimental details see Lichtenthaler and Cuny 22 : yield 87%, melting point [m.p.] 196 °C-198 °C and [α]D 20 = -9.5 (c = 1.1, CHCl3).
1H NMR (500 MHz, CDCl3) δ 1.15-1.45 (m, 4H, 6 Ha, 7 Ha, 8 Ha, 9 Ha), 1.71 (pd, 2H, 7-He, 8-He), 1.82 and 1.88 (each 1H, pd, 6-He, 9-He), 3.89 (m, 2H, 5a-H, 9a-H), 4.11 (ddd, 1H, 2H), 4.47 (dd, 1H, 2′-HA), 4.68 (dd, 1H, 2′-HB), 4.92 (s, 1H, 10a-H), 5.08 (s, 1H, 4a-OH), 5.27 (d, 1H, 4H), 5.87 (pt, 1H, 3H), 7.33, 7.34, and 7.41 (each 2H-pt, aromatic meta-H, in part overlapped), 7.48, 7.50, and 7.54 (each 1 H-m, aromatic para-H, in part overlapped), 7.92, 7.98, and 8.06 (each 2H-dd, aromatic ortho-H); J 2,2′A = 4.7, J 2,2′B = 3.0, J 2′gem = 12.1, J 2,3 = 10.0, J 3,4 = 9.8, aromatic Jortho,meta = 8.5, Jortho,para = 1.5, Jmeta,para = 7.7 Hz.
13C NMR (125.8 MHz, CDCl3) δ 24.25 (C-7, C-8, overlapped), 29.69, 29.99 (C-6*, C-9*), 63.23 (C-2′), 68.08 (C-3), 72.03 (C-2), 72.37, 72.70 (C-5a**, C-9a**), 78.97 (C-4), 91.99 (C-4a), 96.35 (C-10a), 128.45, 128.54, 128.56 (aromatic meta-C), 129.85, 129.92, 130.39 aromatic ortho-C), 133.17, 133.55, 133.92 (aromatic para-C), 165.39, 166.29, 168.44 (3 C6H5 CO); *,** shifts could be interchanged.
Silver carbonate in tetrahydrofuran
A mixture of (1R,2R)-trans-1,2-cycohexanediol (116 mg, 1.00 mmol) and silver carbonate (276 mg, 1.00 mmol) in anhydrous tetrahydrofuran (10 mL) was stirred in the presence of a freshly annealed molecular sieve 4 Å (2 g) at room temperature for 15 minutes. Ulosylbromide
(+)-(2R,3R,3′S,4aR,4′R,5′R,8aR)-3,3′,4′-Tris-Benzoyloxy-5′-(Benzoyloxy)methyl-Octahydro-3H,3′H-Spiro[benzo[b][1,4]dioxine-2,2′-Furan (16)
Starting material 12
Benzoic acid anhydride (2.26 g, 10.00 mmol) and aqueous perchloric acid (70%, 50 µL, 0.58 mmol) were added to a stirred solution of
C40H36O11 (692.7): calcd. C 69.36, H 5.24; found C 69.64, H 5.26.
MS (FD, 15 mA): m/z = 692 (100%, M+).
1H NMR (300 MHz, CDCl3) δ 1.2-2.1 (m, 8H, 5 H2, 6 H2, 7 H2, 8 H2), 3.84* and 4.00* (each 1 H-m, 4a-H and 8a-H), 4.62 (ddd, 1H, 5′-H), 4.79 (1H-dd, 6′-HA), 4.84 (1H-dd, 6′-HB), 5.53 (pd, 1H, 4′ -H), 5.79 (ps, 1H, 3′ -H), 6.45 (s, 1H, 3H), 7.1-8.2 (m, 20H, 4 C6 H 5CO), J 3′,4′ < 0.3, J 4′,5′ = 4.2, J 5′,6′ HA = 4.0, J 5′,6′ HB = 3.8, J 6′gem = 12.1 Hz; *shifts could be interchanged.
13C NMR (75.5 MHz, CDCl3) δ 24.1 (C-6 and C-7, overlapped), 29.5*, 29.9* (C-5, C-8), 63.6 (CH2OBz), 72.4**, 73.0** (C-4a, C-8a), 81.0 (C-3′), 82.0 (C-5′), 89.2 (C-3), 103.8 (C-2), 164.5 (2 C6H5 CO), 165.6, 166.2 (each C6H5 CO); *, ** shifts could be interchanged.
Starting material 17
Benzoic acid anhydride (4.30 g, 19.00 mmol) and aqueous perchloric acid (70%, 100 µL, 1.16 mmol) were added to a stirred solution of
(−)-(2S,3S,3′S,4aS,4′R,5′R,8aS)-3,3′,4′-Tris-Benzoyloxy-5′-(Benzoyloxy)methyl-Octahydro-3H,3′H-Spiro[benzo[b][1,4]dioxine-2,2′-Furan (29 )
Benzoic acid anhydride (4.30 g, 19.00 mmol) and aqueous perchloric acid (70%, 100 µL, 1.16 mmol) were added to a stirred solution of pyran-dioxane-cyclohexane tricycle
C40H36O11 (692.7): calcd. C 69.36, H 5.24; found C 69.58, H 5.31.
MS (FD, 15 mA): m/z = 692 (100%, M+).
1H NMR (300 MHz, CDCl3) δ 1.1-2.0 (m, 8H, 5 H2, 6 H2, 7 H2, 8 H2), 3.78* and 4.05* (each 1 H-m, 4a-H and 8a-H), 4.64 (ddd, 1H, 5′-H), 4.75 (dd, 1H, 6′-HA), 4.81 (dd, 1H, 6′-HB), 5.88 (dd, 1H, 4′-H), 5.96 (d, 1H, 3′-H), 6.33 (s, 1H, 3H), 6.9-8.2 (m, 20H, 4 C6 H 5CO); J 3′,4′ =6.9, J 4′,5′ =5.3, J 5′,6′ HA = 5.1, J 5′,6′ HB = 6.4, J 6′gem = 11.6. Hz; *shifts could be interchanged.
13C NMR (75.5 MHz, CDCl3) δ 24.0 (C-6 and C-7, overlapped), 29.5*, 29.8* (C-5, C-8), 65.4 (CH2OBz), 72.7**, 73.2** (C-4a, C-8a), 75.6 (C-3′), 78.0 (C-4′), 79.1 (C-5′), 90.8 (C-3), 100.2 (C-2), 164.5, 164.8, 165.9, 166.2 (each C6H5 CO); *, ** shifts could be interchanged.
1,2,3,4,6-Penta-O-Benzoyl-d -Glucopyranose (30 )
In a 3-necked round-bottom flask with a mechanical stirrer and dropping funnel, benzoyl chloride (150 mL, 1.2 mol) was added to a mixture of anhydrous
1H NMR (500 MHz, CDCl3) main α-anomer: δ 4.52 (dd, 1H, 6-HA), 4.65 (m, 2H, 5H, 6-HB), 5.72 (dd, 1H, 2H), 5.89 (pt, 1H, 4H), 6.35 (pt, 1H, 3H), 6.89 (d, 1H, 1H), 7.25-8.18 (m, 25H, 5 C6 H 5CO); J 1,2 = 3.7, J 2,3 = 10.2, J 3,4 = 9.7, J 4,5 = 9.7, J 5,6A = 5.6, J 6gem = 13.1 Hz.
13C NMR (125.8 MHz, CDCl3) δ 62.62 (C-6), 69.01 (C-4), 70.58*, 70.62*, 70.64* (C-2, C-3, C-5), 90.18 (C-1), 128.1-130.4 (aromatic meta and para-C), 133.1-134.2 (aromatic ortho-C), 164.50, 165.25, 165.46, 166.02, 166.18 (5 C6H5 CO); *shifts could be interchanged.
Minor β-anomer: δ 4.44 (ddd, 1H, 5H), 4.54 (dd, 1H, 6-HA), 4.69 (dd, 1H, 6-HB), 5.86 (pt, 1H, 4H), 5.89 (pt, 1H, 2H hidden by 4H of α-anomer), 6.08 (pt, 1H, 3H), 6.34 (d, 1H, 1H), 7.25-8.18 (m, 25H, 5 C6 H 5CO); J 1,2 = 8.0, J 2,3 = 9.5, J 3,4 = 9.5, J 4,5 = 9.8, J 5,6A = 4.8, J 5,6B = 3.0, J 6gem = 12.4 Hz.
13C NMR (125.8 MHz, CDCl3) δ 62.83 (C-6), 69.25 (C-4), 71.02 (C-2), 72.98 (C-3), 73.63 (C-5), 92.85 (C-1), 128.1‐130.4 (aromatic meta and para-C overlapped of α-anomer-C), 133.1-134.2 (aromatic ortho-C overlapped of α-anomer-C), 164.2-166.4 (5 C6H5 CO overlapped of α-anomer-C).
2,3,4,6-Tetra-O-Benzoyl-α-d -Glucopyranosyl Bromide (31 )
In a 3-necked round-bottom flask with a mechanical stirrer and dropping funnel, hydrobromic acid (HBr) 1.2 M solution in glacial acetic acid (80 mL, 0.5 mol) was added under stirring at room temperature to a solution of 1,2,3,4,6-penta-O-benzoyl-
2,3,4,6-Tetra-O-Benzoyl-5-Anhydro1-d -Arabino-Hex-1-Enitol (Tetra-O-Benzoyl-2-Hydroxy-d -Glucal) (32 )
In a round-bottom flask with a mechanical stirrer, 33.2 g (0.05 mol) of the above prepared 2,3,4,6-tetra-O-benzoyl-α-
1H NMR (500 MHz, CDCl3) δ 4.73 (dd, 1H, 6-HA), 4.90 (ddd, 1H, 5H, overlapped with 6-HB), 4.91 (dd, 1H, 6-HB, overlapped with 5H), 5.85 (pt, 1H, 4H), 6.12 (d, 1H, 3H), 6.99 (s, 1H, 1H), aromatic meta-H: 7.35-7.44 (m, 6H), and 7.47 (pt, 2H), aromatic para-H: 7.50-7.62 (m, 4H), aromatic ortho-H: 7.98 (dd, 2H), 8.03 (m, 4H), and 8.12 (dd, 1H); J 3,4 = 3.7, J 4,5 = 4.0, J 5,6A = 3.5, J 5,6B = 7.2, J 6gem = 10.8, aromatic Jortho,meta = 8.3, Jortho,para = 1.1, Jmeta,para = 8.0 Hz.
13C NMR (125.8 MHz, CDCl3) δ 61.66 (C-6), 66.73 (C-3), 68.37 (C-4), 73.97 (C-5), 128.53, 128.59 (2s), 128.67 (aromatic meta-C), 129.85, 129.94, 130.20 (2s) (aromatic para-C), 133.34, 133.50, 133.64, 133.74 (aromatic ortho-C), 139.88 (C-1), 165.15, 165.47, 165.61, 166.18 (4 C6H5 CO).
Tri-O-Benzoyl-α-d -Arabino-Hexopyranos-2-Ulosylbromide (11 )
In a round-bottom flask with a mechanical stirrer and drying tube, filled up with calcium chloride (CaCl2), 2,3,4,6-tetra-O-benzoyl-2-hydroxy-
1H NMR (500 MHz, CDCl3) δ 4.56 (dd, 1H, 6-HA), 4.76 (dd, 1H, 6-HB), 4.93 (ddd, 1H, 5H), 6.04 (pt, 1H, 4H), 6.51 (s, 1H, 1H), 6.53 (d, 1H, 3H), 7.41, 7.42, and 7.45 (each 2H-pt, aromatic meta-H, in part overlapped), 7.54, 7.56, and 7.59 (each 1 H-m, aromatic para-H, in part overlapped), 8.00, 8.02, and 8.05 (each 2H-dd, aromatic ortho-H); J 3,4 = 10.4, J 4,5 = 10.4, J 5,6A = 4.5, J 5,6B = 2.4, J 6gem = 12.6, aromatic J ortho,meta = 8.0, J ortho,para = 0.8, J meta,para = 7.8 Hz.
13C NMR (125.8 MHz, CDCl3) δ 61.72 (C-6), 68.45 (C-4), 73.12 (C-5), 73.20 (C-3), 83.19 (C-1), 128.63, 128.66, 128.75 (aromatic meta-C), 129.96, 130.10, 130.22 (aromatic ortho-C), 133.52, 133.87 ,134.05 (aromatic para-C), 164.66, 165.25, 166.02 (3 C6H5 CO).
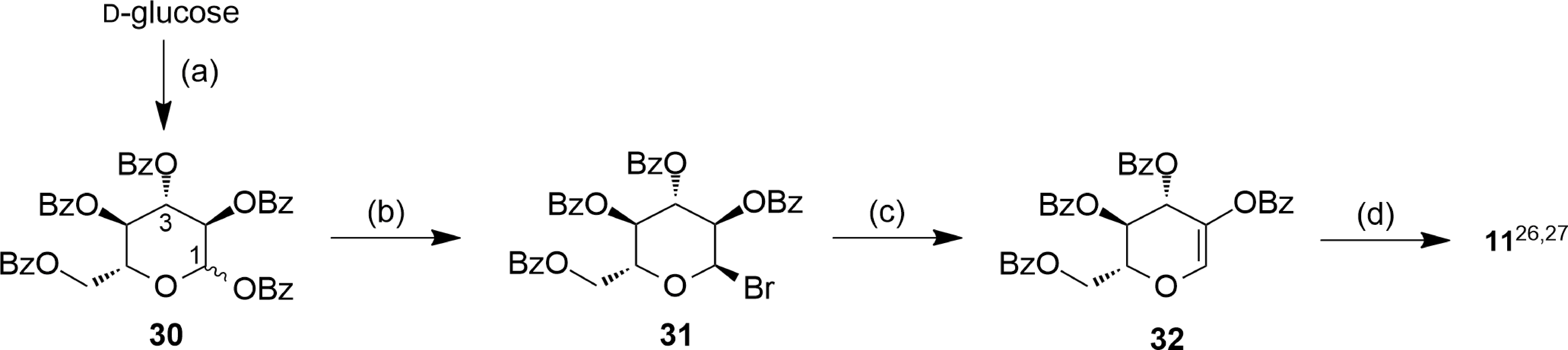
Large-scale synthesis of ulosylbromide 11 from
Footnotes
Acknowledgment
The author thanks Prof. Dr. M Reggelin for the opportunity to work in his group.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.