
Abstract
Spiroacetals are the central structural core element of numerous natural products and are essential for their biological activity. A typical structural representative of a spiroacetal is the bicyclic 1,6-dioxaspiro[4.5]decane ring system. It represents the complete or partial structure of many biologically potent natural products such as the Paravespula pheromone
Spiroacetals are the central structural core element in a multitude of natural products of insect, marine, bacterial, or plant origin. 1 -6 A typical example of an organism producing a spiroacetal compound is the wasp Paravespula vulgaris, a social insect constructing and living in communal nests. The wasps emit an aggregation pheromone with spiroacetal molecular structure (Figure 1) for the purpose of mating and overwhelming predators.

(a) The wasp Paravespula vulgaris taking water on duckweed Lemna minor. (b) Paravespula nest attached to a concrete ceiling and superimposed in yellow the species-specific spiroacetal molecular structure of the aggregation pheromone produced by these insects. Photos were taken by the author in his garden and garage, summer 2019, Seeheim, Germany.
This aggregation pheromone is the minor bouquet component and a mixture of 2 diasteriomers of 2-methyl-1,6-dioxaspiro[4.5]-decane with either cis or trans geometry in the furan ring of the methyl group and the pyran oxygen atom
7
(

Typical spiroketal natural products with 1,6-dioxaspiro[4.5]decane structural motif (highlighted in red): Paravespula pheromone 1, (+)-monensin A 2, (−)-berkelic acid 3, (+)-spirastrellolide F methyl ester 4, and (−)-calyculin A 5. Whilst the 1,6-dioxaspiro[4.5]decane motifs (R)-
Biological studies disclosed that the spiroacetal framework is essential for biological activity in these types of natural compounds. 35 -40 Given their diverse biological activity, Zinzalla et al 41 designed and synthesized spiroketal derivatives, leading to a collection of new small molecules for biological evaluation as orally bioavailable lead compounds. The use of spirocyclic scaffolds in drug discovery has been reviewed. 42 With regard to drug design, in 2017, Scheepstra et al 43 published a paper entitled ”Designed Spiroketal Protein Modulation”, describing that spiroketals are arguably well-suited for 2 approaches: (1) diversity-oriented synthesis and (2) biology-oriented synthesis.
The 1,6-dioxaspiro[4.5]decane ring system is found in nature with either spiro (R)- or spiro (S)-configuration (structures (R)-
Syntheses
The chemical elaboration of the 1,6,9-trioxaspiro[4.5]decane ring system is difficult and only a few racemic approaches are known. These include the electrolysis of mono furfuryl ethers,
44,45
a carbon–carbon bond formation reaction of a diazoketone
46
and the selective reduction of analogs of the secondary metabolite and antifeedant tonghaosu.
47,48
A newer US patent
49
describes the cyclization of keto diols with 1 stereogenic center, which yields derivatives of
In the early 1980s, we developed a practical method for the conversion of the 2 literature-known
Here we describe for the first time the glycosylation of the 6-deoxyulosyl bromide
Derivatives with Spiroacetal Motif (R)-7
The stereoselective synthesis of compounds with the spiroacetal motif (R)-
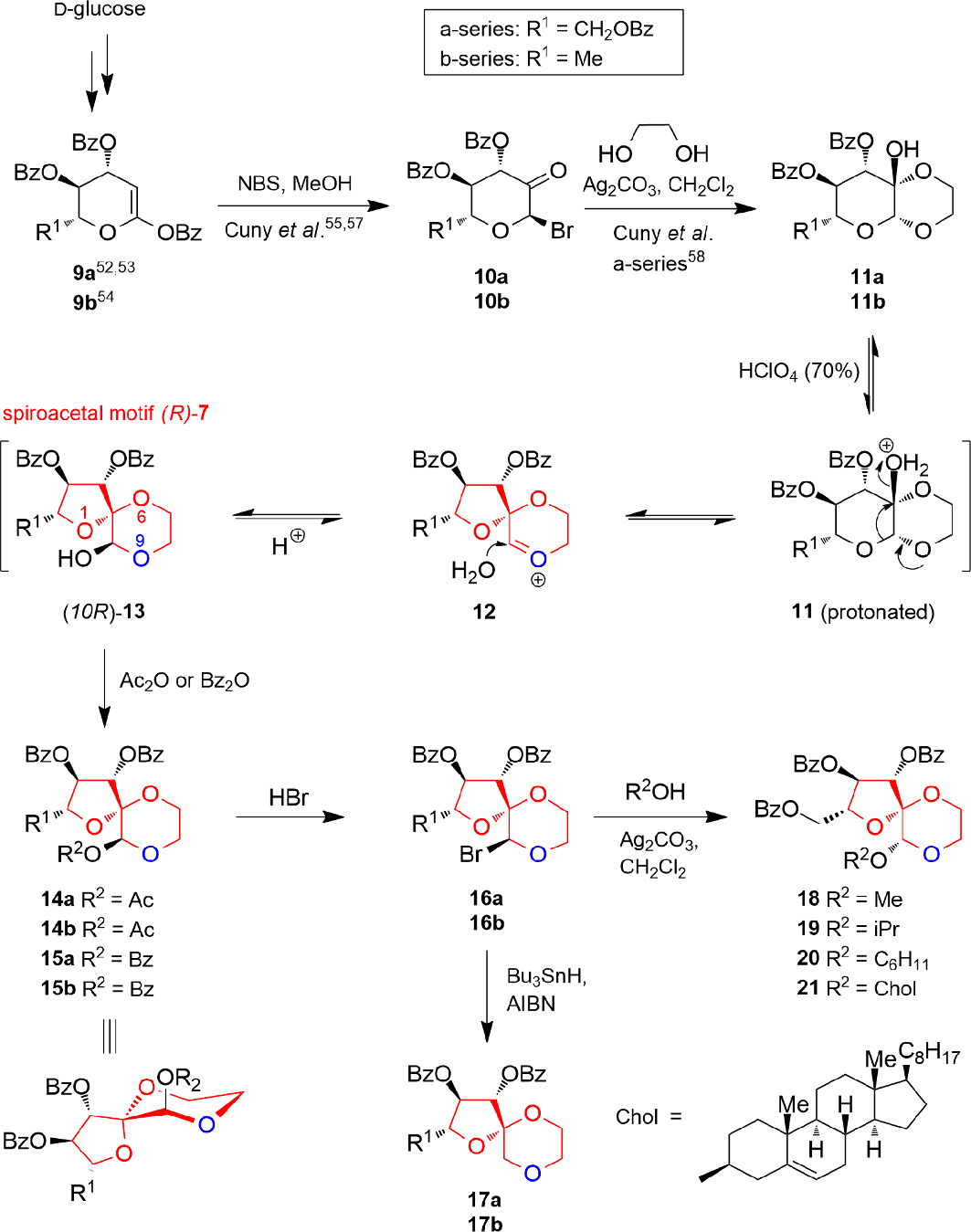
Conversion of the
The reaction of the 6-deoxy-ulosyl bromide
As shown by a comparison experiment, no appreciable conversion occurred by the reaction of
Subsequent bromination of
The synthesized compounds possess the (5R)-1,6,9-tri-oxaspiro[4.5]decane framework (R)-
Derivative with Spiroacetal Motif (S)-7
The stereospecific conversion of the pyranodioxane
The pyranodioxane
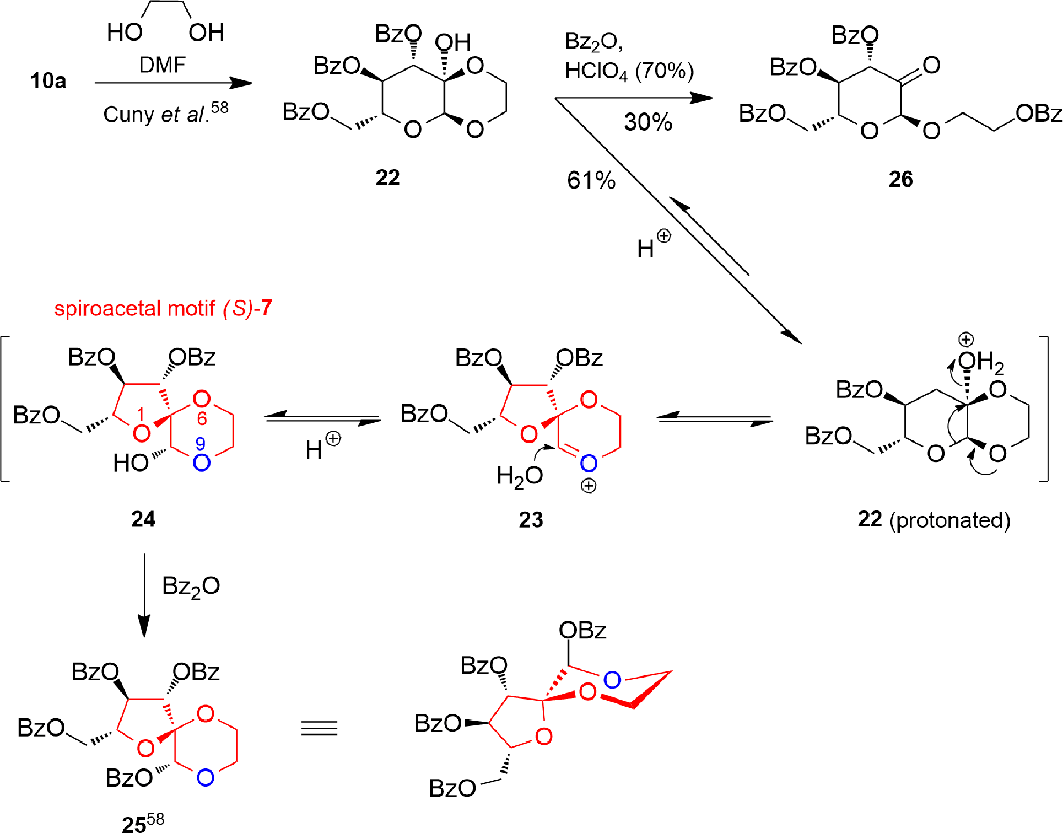
The examination of the acid catalyzed ring contraction of the pyranodioxane 22 revealed that in the presence of benzoic acid anhydride, not only the required 25 was formed but also the undesired open-chain side product 26. Protonated starting material 22 and structures 23 and 24 are intermediates on the reaction path to 25. The starting material 22 is readily obtainable from
The synthesized 1,6,9-tri-oxaspiro[4.5]decane derivative
Structural Elucidation
The configuration and conformation of the aforementioned 1,6,9-tri-oxaspiro[4.5]decanes with either (R)- or (S)-spiro framework could be resolved on the basis of 1H and 13C NMR data.
Furan Portion
The relevant proton chemical shifts and coupling constants of the ring protons in the tetrahydrofuran portion are listed in Table 1.
Selected Proton Chemical Shifts and 1H-1H Coupling Constants of 1,6,9-Trioxaspiro[4.5]decanes in the Tetrahydrofuran Portion (300 MHz in CDCl3, δ in ppm, J in Hz) a .
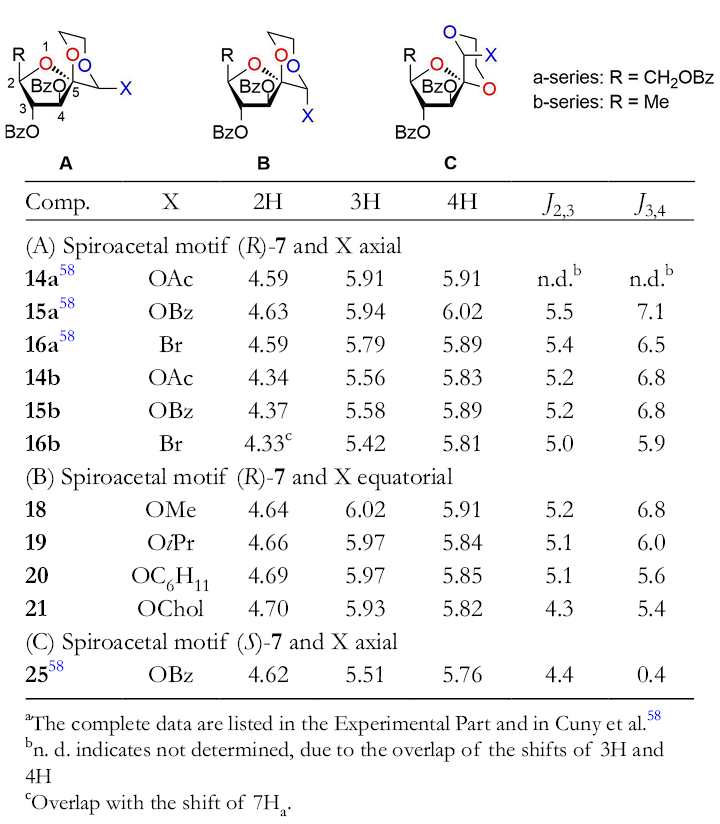

aThe complete data are listed in the Experimental Part and in Cuny et al. 58
bn. d. indicates not determined, due to the overlap of the shifts of 3H and 4H
cOverlap with the shift of 7Ha.
The chemical shifts for 2H, 3H, and 4H of compounds with the spiroacetal motif (R)-
The same result was also found by Lemieux and Nagarajan
59
in the hexaacetate of di-

Molecular geometry and 1H-1H coupling constants of the hexaacetate of di-
1,4-Dioxane Portion
The chemical shifts of the ring protons in the dioxane portion are listed in Table 2. Their axial/equatorial orientation could be easily distinguished by the width of their chemical shifts.
Selected Proton Chemical Shifts of 1,6,9-Trioxaspiro[4.5]decanes in the 1,4-Dioxane Portion with Either (R)- or (S)-Configuration (300 MHz in CDCl3, δ in ppm) a .


aThe multiplicity of the shifts and the coupling constants are listed in the section Experimental and in Cuny et al. 58
bOverlap with the shifts of 2H.
Due to the large geminal and axial coupling constants, 7Ha and 8Ha possess broad ddd-shifts whereas 7He and 8He are characterized by narrow pdd-shifts, because only one large geminal coupling constant is found.
7Ha and 8Ha could be further differentiated on the basis of the strong downfield shift for 8Ha (4.45–4.54 ppm vs 4.12–4.33 for 7Ha) in compounds with an axial substituent at C-10 (A and C in Table 2). Responsible for this extensive effect is the 1,3-diaxial interaction of the electronegative substituent X at C-10 with 8Ha.
The determination of 8Ha in compounds possessing an equatorial substituent X at C-10 was substantiated by the observed NOE interactions of 10 H → 8 Ha (B in Table 2).
Due to the relatively long distance of 5 bonds to the substituent at C-10, the values of the chemical shift for the 2 geminal protons 7Ha (4.12-4.33 ppm) and 7He (3.45-3.70 ppm) are approximately of the same magnitude in all cases (A, B, and C in Table 2). The same result is obtained for 8He (3.67-3.88 ppm) possessing a distance of 4 bonds.
13C NMR Spectral Data
The 13C NMR spectral data of the synthesized 1,6,9-trioxaspiro[4.5]decanes are listed in Table 3. For each of these compounds, the values of the chemical shifts correlate well. As expected, notable differences are found for the carbon atom C-10, because it possesses different substituents. Thus, the values for the chemical shifts for
Selected 13C Nuclear Magnetic Resonance Data a of 1,6,9-Trioxaspiro[4.5]decanes (75.5 MHz in CDCl3, δ in ppm).
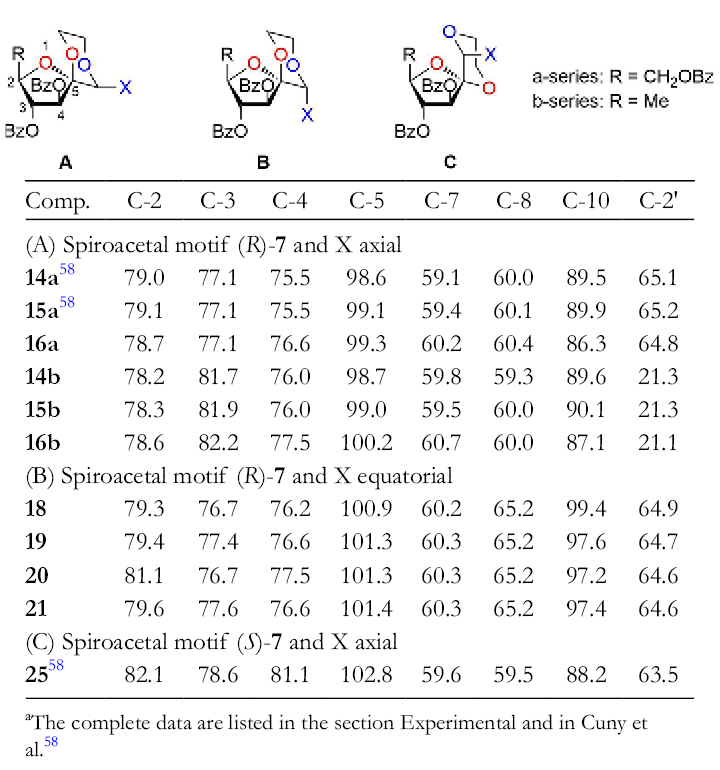

aThe complete data are listed in the section Experimental and in Cuny et al. 58
Concluding Remarks
The 1,6-dioxaspiro[4.5]decane motifs (R)-
The 6-deoxy-pyranodioxane
The synthesized compounds possess the (5R)-1,6,9-tri-oxaspiro[4.5]decane framework (R)-
The synthesis of (5S)-1,6,9-trioxaspiro[4.5]decanes with opposite chirality is also possible.
58
To further clarify the reaction process, we studied the formation of the (5S)-1,6,9-trioxaspiro[4.5]decane
The 1H and 13C NMR data of 1,6,9-tri-oxaspiro[4.5]decanes that are published and described herein are summarized in Tables 1
-3. On the basis of these data, together with an X-ray analysis of compound
The axial or equatorial arrangement of the substituent at C-10 could be established from the chemical shift value of the 8Ha dioxane proton, which is 4.45-4.50 ppm for an axial arrangement and 3.82-3.84 ppm for an equatorial configuration (see Table 2). The influence of the C-10 substituent on the values of the chemical shifts of the tetrahydrofuran ring protons is not significant because it is relatively far away.
The synthesized 1,6,9-tri-oxaspiro[4.5]decane motifs
Experimental
TLC was performed on POLYGRAM SILG/UV254 (Macherey Nagel & Co.). Preparative chromatographic separations were carried out on columns with Merck silica gel 60 (15–40 µm) and Merck precoated silicagel plates 60 F254, 20 × 20 cm, 0.25 mm. Melting points (m.p.) were determined on a Bock-Monoskop VS or a Büchi SMP-20 and are uncorrected. Specific optical rotations were determined on a Perkin-Elmer Polarimeter 241 in 1 dm cuvettes at a wavelength of 589 nm. NMR spectra were measured on a Bruker WM 300 spectrometer at 303 K using TMS as an internal reference. The abbreviation of the multiplicities of the shifts is indicated as s for singlet, d for doublet, t for triplet, q for quartet, sxt for sextet, oct for octet, m for multiplet, br for broad, and p for pseudo. Mass spectra were run on a MAT 311 mass spectrometer (Varian). Elemental analyses were performed on a Perkin Elmer 240 Elementar Analyser.
(−)-(4aR,6R,7R,8S,8aS)-7,8-Bis-Benzoyloxy-6-Methyl-8a-Hydroxy-1,4,5-Trioxa-Cis-Decaline (11b)
A mixture of silver carbonate (662 mg, 2.40 mmol), ethylene glycol (134 µL, 149 mg, 2.40 mmol) in water-free dichloromethane (25 mL) was stirred in the presence of a freshly annealed molecular sieve 4 Å (2 g) at room temperature for 15 minutes. 6-Deoxy-ulosyl bromide
C22H22O8 (414.4): calcd. C 63.76, H 5.35; found C 63.85, H 5.31.
MS (FD, 20 mA): m/z = 415 (100%, M + 1).
1H NMR (300 MHz, CDCl3) δ 1.38 (d, 3H, 6-CH 3), 3.57, 4.33 (each 2 H-m, 2H2, 3H2), 3.82 (dq, 1 H, 6 H), 4.71 (s, 1 H, 4a H), 5.12 (d, 1 H, 8 H), 5.29 (s, 1 H, OH), 5.51 (dd, 1 H, 7 H), 7.2-8.2 (m, 10 H, 2C6 H 5CO). J 6,CH3 = 6.1, J 6,7 = 9.8, J 7,8 = 9.7 Hz.
13C NMR (75.5 MHz, CDCl3) δ 17.5 (CH3), 59.1, 59.3 (C-2, C-3), 70.0 (C-6), 72.2 (C-7), 79.5 (C-8), 91.1 (C-8a), 95.2 (C-4a), 128-134 (2 C 6H5), 165.5, 168.6 (2 C6H5 CO).
Diastereomeric mixture of (−)-(2R,3R,4S,5S,10R,S)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Hydroxy-1,6,9-Trioxaspiro[4.5]decane (10R,S) (13a )
Aqueous perchloric acid (70 %, 0.8 mL, 9.3 mmol) was added to a stirred solution of the 1,4,5-trioxa-cis-decaline
C29H26O10 (534.5): calcd. C 65.16, H 4.90; found C 65.31, H 5.02.
MS (FD, 20 mA): m/z = 533 (25 %, M+ − 1), 412 (100%, M+–C6H5COOH).
Main (10S)-
1H NMR (300 MHz, CDCl3) δ: 3.47 (dd, 1 H, 8 He), 3.84 (m, 2 H, 7 Ha, 7 He), 4.10 (m, 1 H, 8 Ha) 4.66 (m, 2 H, 2 H, 2′ HA), 4.79 (m, 1 H, 2′ HB), 5.00 (s, 1 H, 10 H, after treatment with D2O), 5.01 (br s, 1 H, OH), 5.91 (d, 1 H, 4 H), 5.97 (dd, 1 H, 3 H); J 2,3 = 4.4, J 3,4 = 5.9, J 8gem = 11.8, J 7a,8e = 2.1 Hz, all other shifts are overlapped.
13C NMR (75.5 MHz, CDCl3) δ: 60.1 (C-7), 64.7 (CH2OBz), 65.3 C-8), 76.3/C-4), 77.5 (C-3), 80.1 (C-2), 91.8 (C-10), 102.0 (C-5), 128.0–134.0 (3 C 6H5CO), 165.6, 165.8, 166.2 (3 C6H5 CO).
Minor (10R)-
1H NMR (300 MHz, CDCl3) δ: 5.02 (s, 1 H, 10 H, after treatment with D2O), 5.04 (br s, 1 H, OH), 5.83 (d, 1 H, 4 H), 5.87 (dd, 1 H, 3 H); J 2,3 = 3.9, J 3,4 = 5.1 Hz, all other shifts are overlapped.
13C NMR (75.5 MHz, CDCl3) δ: 57.2 (C-8), 61.0 C-7), 64.7 (CH2OBz), 77.5 (C-4), 77.8 (C-3), 79.7 (C-2), 91.5 (C-10), 100.8 (C-5), 128.0-134.0 (3 C 6H5CO), 165.6, 165.8, 166.2 (3 C6H5 CO).
(−)-(2R,3R,4S,5S,10S)-10-Acetyloxy-3,4-Bis-Benzoyloxy-2-Methyl-1,6,9-Trioxaspiro[4.5]decane (14b)
Acetic acid anhydride (13.7 mL, 144.8 mmol) and aqueous perchloric acid (70%, 0.5 mL, 5.8 mmol) were added to a stirred solution of the 1,4,5-trioxa-cis-decaline
C24H24O9 (456.4): calcd. C 63.15, H 5.30; found C 63.20, H 5.29.
MS (FD, 20 mA): m/z = 456 (100%, M+).
1H NMR (300 MHz, CDCl3) δ: 1.65 (d, 3 H, 2-CH 3), 1.86 (s, 3 H, CH 3CO), 3.59 (pdd, 1 H, 7 He), 3.74 (pdd, 1 H, 8 He), 4.14 (ddd, 1 H, 7 Ha), 4.34 (psxt, 1 H, 2 H), 4.46 (ddd, 1 H, 8 Ha), 5.56 (dd, 1 H, 3 H), 5.83 (d, 1 H, 4 H), 5.93 (s, 1 H, 10 H), 7.3-8.2 (m, 10 H, 2 C6 H 5CO); J 2,Me = 6.6, J 2,3 = 5.2, J 3,4 = 6.8, J 7gem = 11.7, J 7a,8a = 12.2, J 7a,8e = 3.0, J 7e,8a = 3.1, J 7e,8e = 0, J 8gem = 11.6 Hz.
13C NMR (75.5 MHz, CDCl3) δ: 20.8 (CH3CO), 128.0-134.0 (2 C 6H5CO), 165.3, 166.0 (2 C6H5 CO), 169.4 (CH3 CO). For all other shifts see Table 3.
(−)-(2R,3R,4S,5S,10S)-3,4,10-Tris-Benzoyloxy-2-Methyl-1,6,9-Trioxaspiro[4.5]decane (15b)
Benzoic acid anhydride (2.72 g, 12.0 mmol) and aqueous perchloric acid (70%, 50 µL, 0.6 mmol) were added to a stirred solution of the 1,4,5-trioxa-cis-decaline
C29H26O9 (518.5): calcd. C 67.17, H 5.05; found C 67.25, H 5.12.
MS (FD, 20 mA): m/z = 518 (100%, M+).
1H NMR (300 MHz, CDCl3) δ: 1.68 (d, 3 H, 2-CH 3), 3.64 (pdd, 1 H, 7 He), 3.81 (pdd, 1 H, 8 He), 4.24 (ddd, 1 H, 7 Ha), 4.37 (psxt, 1 H, 2 H), 4.54 (ddd, 1 H, 8 Ha), 5.58 (dd, 1 H, 3 H), 5.89 (d, 1 H, 4 H), 6.20 (s, 1 H, 10 H), 6.9-8.2 (m, 15 H, 3 C6 H 5CO); J 2,Me = 6.5, J 2,3 = 5.2, J 3,4 = 6.8, J 7gem = 11.7, J 7a,8a = 12.2, J 7a,8e = 2.8, J 7e,8a = 2.7, J 7e,8e = 0, J 8gem = 11.6 Hz.
13C NMR (75.5 MHz, CDCl3) δ: 164.5, 164.8, 166.1 (3 C6H5 CO). For all other shifts see Table 3.
(−)-(2R,3R,4S,5S,10S)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Bromo-1,6,9-Trioxaspiro[4.5]decane (16a)
Preparation, physical and spectral data see Cuny et al.. 58
13C NMR (75.5 MHz, CDCl3) δ: 124.2-144.2 (3 C 6H5CO) 165.2, 165.6, 166.2 (3 C6H5 CO). For all other shifts see Table 3.
(−)-(2R,3R,4S,5S,10S)-3,4-Bis-Benzoyloxy-10-Bromo-2-Methyl-1,6,9-Trioxaspiro[4.5]decane (16b)
(A) Bromination of acetate
C22H21BrO7 (477.3): calcd. C 55.36, H 4.22; found C 55.54, H 4.31.
1H NMR (300 MHz, CDCl3) δ 1.61 (d, 3 H, 2-CH 3), 3.70 (pdd, 1 H, 7 He), 3.83 (pdd, 1 H, 8 He), 4.33 (m, 2 H, 2 H and 7 Ha), 4.50 (ddd, 1 H, 8 Ha), 5.42 (dd, 1 H, 3 H), 5.81 (d, 1 H, 4 H), 6.23 (s, 1 H, 10 H), 7.3-8.2 (m, 10 H, 2 C6 H 5CO); J 2,Me = 6.5 J 2,3 = 5.0, J 3,4 = 5.9, J 7gem = 11.7, J 7a,8a = 12.2, J 7a,8e = 3.1, J 7e,8a = 2.9, J 7e,8e = 0, J 8gem = 11.5 Hz.
13C NMR (75.5 MHz, CDCl3) δ: 128.0-134.0 (2 C 6H5CO) 165.2, 166.0 (2 C6H5 CO). For all other shifts see Table 3.
(B) Bromination of benzoate
(−)-(2R,3R,4S,5R)-3,4-Bis-Benzoyloxy-2-Methyl-1,6,9-Trioxaspiro[4.5]decane (17b)
Azobisisobutyronitrile (AIBN) (25 mg, 0.15 mmol) and tributyltin hydride (350 µL, 1.33 mmol) were successively added to a solution of bromide
MS (FD, 20 mA): m/z = 398 (80 %, M+), 397 (100%, M+ − 1).
1H NMR (300 MHz, CDCl3) δ: 1.64 (d, 3 H, 2-CH 3), 3.60-3.80 (m, 3 H, 7 He, 8 Ha and 8 He), 3,79 (d, 1 H, 10 Ha), 3,85 (d, 1 H, 10-He), 4.20-4.40 (m, 2 H, 2 H and 7 Ha), 5.50 (d, 1 H, 4 H), 5.65 (dd, 1 H, 3 H), 7.3-8.2 (m, 10 H, 2 C6 H 5CO); J 2,Me = 6.5, J 2,3 = 4.8, J 3,4 = 6.3, J 10gem = 11.8 Hz.
(−)-(2R,3R,4S,5S,10S)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Methoxy-1,6,9-Trioxaspiro[4.5]decane (18)
Bromide
C30H28O10 (548.5): calcd. C 65.69, H 5.15; found C 65.48, H 5.06.
MS (FD, 20 mA): m/z = 548 (100 %, M+).
1H NMR (300 MHz, CDCl3) δ: 3.46 (pdd, 1 H, 7 He), 3.68 (s, 3 H, OCH 3), 3.82 (ddd, 1 H, 8 Ha), 3.88 (pdd, 1 H, 8 He), 4.19 (ddd, 1 H, 7 Ha), 4.64 (s, 1 H, 10 H and m, 1 H, 2 H), 4.65 (dd, 1 H, HA of CH2OBz), 4.77 (dd, 1 H, HB of CH2OBz), 5.91 (d, 1 H, 4 H), 6.02 (dd, 1 H, 3 H), 7.3-8.2 (m, 15 H, 3 C6 H 5CO); J 2,HA = 5.4, J 2,HB = 6.3, J HA, HB = 13.6, J 2,3 = 5.2, J 3,4 = 6.8, J 7gem = 11.6, J 7a,8a = 11.4, J 7a,8e = 3.5, J 7e,8a = 2.3, J 7e,8e = 0, J 8gem = 11.8 Hz.
NOE interactions 10 H (4.64) → OCH3 (3.68), 8 Ha (3.82) and 4 H (5.91 ppm).
13C NMR (75.5 MHz, CDCl3) δ: 57.4 OCH3), 128.0-134.0 (3 C 6H5CO), 165.8, 165.9, 166.2 (3 C6H5 CO). For all other shifts see Table 3.
(−)-(2R,3R,4S,5S,10R)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Isopropyloxy-1,6,9-Trioxaspiro[4.5]decane (19)
Bromide
C30H28O10 (576.6): calcd. C 66.66, H 5.59; found C 66.79, H 5.45.
MS (FD, 20 mA): m/z = 576 (100 %, M+).
1H NMR (300 MHz, CDCl3) δ: 1.32 (d, 6 H, 2 isopropyl-CH 3), 3.46 (m, 1 H, 7 He), 3.83 (m, 2 H, 8 Ha and 8 He), 4.11 (q, 1 H, isopropyl-CH), 4.18 (m, 1 H, 7 Ha), 4.66 (m. 2 H, 2 H and HA of CH2OBz), 4.73 (dd, 1 H, HB of CH2OBz), 4.81 (s, 1 H, 10 H), 5.84 (d, 1 H, 4 H), 5.97 (dd, 1 H, 3 H), 7.3–8.2 (m, 15 H, 3 C6 H 5CO); J 2,HA = 5.0, J HA, HB = 13.6, J 2,3 = 5.1, J 3,4 = 6.0, J CH,Me = 6.6 Hz, all other shifts are overlapped.
13C NMR (75.5 MHz, CDCl3) isopropyl CH3: δ: 22.1, 23.6; isopropyl CH 72.8; 128.0–134.0 (3 C
6H5CO), 2 × 165.8, 166.2 (3 C6H5
(−)-(2R,3R,4S,5S,10R)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Cycolhexyloxy-1,6,9-Trioxaspiro[4.5]decane (20)
Cyclohexanol (170 mg, 180 µL, 1.67 mmol) was dissolved in anhydrous dichloromethane (50 mL) followed by the addition of silver carbonate (460 mg, 1.67 mmol) and freshly dried molecular sieve 4 Å (2 g). After the mixture was stirred for 15 minutes at room temperature under the absence of light, bromide
C35H36O10 (616.6): calcd. C 68.17, H 5.89; found C 67.65, H 6.01.
MS (FD, 20 mA): m/z = 616 (100 %, M+).
1H NMR (300 MHz, CDCl3) δ: 0.7-2.4 (m, 10 H, 4 cylohexyl-CH 2), 3.46 (pdd, 1 H, 7 He), 3.78 (m, 1 H, cyclohexyl-CH), 3.83 (m, 2 H, 8 Ha and 8 He), 4.19 (m, 1 H, 7 Ha), 4.66 (dd. 2 H, HA of CH2OBz), 4.69 (m, 1 H, 2 H), 4.75 (dd, 1 H, HB of CH2OBz), 4.89 (s, 1 H, 10 H), 5.85 (d, 1 H, 4 H), 5.97 (dd, 1 H, 3 H), 7.3-8.2 (m, 15 H, 3 C6 H 5CO); J 2,HA = 4.8, J 2,HB = 6.4, J HA, HB = 10.4, J 2,3 = 5.1, J 3,4 = 5.6 Hz, all other shifts are overlapped.
NOE interactions 10 H (4.89) → 8 Ha (3.83), 4 H (5.85) and cyclohexyl-CH (3.78 ppm).
13C NMR (75.5 MHz, CDCl3) cyclohexyl CH2: δ: 24.1, 25.5, 31.8, 39.7; cyclohexyl CH 79.4; 128.0–134.0 (3 C 6H5CO), 2 × 165.8, 166.2 (3 C6H5 CO). For all other shifts see Table 3.
(+)-(2R,3R,4S,5S,10R,3′R)-3,4-Bis-Benzoyloxy-2-Benzoyloxymethyl-10-Cholestanyloxy-1,6,9-Trioxaspiro[4.5]decane (21)
(3β,5α)-Cholestane-3-ol (194 mg, 0.50 mmol) was dissolved in anhydrous dichloromethane (10 mL) followed by the addition of silver carbonate (138 mg, 0.50 mmol) and freshly dried molecular sieve 4 Å (2 g). After the mixture was stirred for 15 minutes at room temperature under the absence of light, bromide
C56H72O10 (905.2): calcd. C 74.31, H 8.02; found C 73.84, H 7.96.
MS (FD, 20 mA): m/z = 904 (100 %, M+ − 1).
1H NMR (300 MHz, CDCl3) 1,6,9-trioxaspiro[4.5]decane portion δ: 3.45 (pdd, 1 H, 7 He), 3.83 (m, 2 H, 8 Ha, and 8 He), 4.17 (m, 1 H, 7 Ha), 4.70 (m, 3 H, 2 H, HA and HB of CH2OBz), 4.86 (s, 1 H, 10 H), 5.82 (d, 1 H, 4 H), 5.93 (dd, 1 H, 3 H), 7.2–8.2 (m, 15 H, 3 C6 H 5CO); J 2,3 = 4.3, J 3,4 = 5.4 Hz, all other shifts are overlapped.
Main shifts of cholestane portion δ: 0.65 (s, 3 H, 18 H3), 0.76 (s, 3 H, 19 H3), 0.86 (2 3H-d, 26 H3 and 27 H3), 0.90 (d, 3 H, 21 H3), 3.73 (m, 1 H, 3 H); J 20,21 = 6.5, J 25,26 = 6.6, J 25,27 = 6.6 Hz.
13C NMR (75.5 MHz, CDCl3) steroidal CH3: δ 12.1 (C-18), 12.2 (C-19), 18.6 (C-21), 22.6 (C-26), 22.7 (C-27); steroidal CH2: 21.2, 23.8, 24.2, 28.0, 28.3, 28.7, 29.7, 32.1, 36.2, 36.9, 39.5, 40.0; steroidal CH: 28.0, 35.5, 35.8, 44.9, 54.3, 56.3, 56.5, 79.4; steroidal C: 35.5, 42.6; 128.0–134.0 (3 C 6H5CO), 2 × 165.8, 166.2 (3 C6H5 CO). For all other shifts see Table 3.
(+)-(2R,3R,4S,5R,10R)-3,4,10-Tris-Benzoyloxy-2-Benzoyloxymethyl-1,6,9-Trioxaspiro[4.5]decane (25 ) and (+)-1-(2′-Benzoyloxy)-Ethyl-3,4,6-Tri-O-Benzoyl-a-d -Arabino-Hexopyranos-2-ulosid (26)
Benzoic acid anhydride (1.81 g, 8.00 mmol) and perchloric acid (70%) (50 µL, 0.60 mmol) were added to a solution of pyranodioxane
The remaining residue was filtered through a silica gel column (15 × 2 cm, n-hexane/diethyl ether 3:1) to remove excessive benzoic acid anhydride and polar impurities. The eluate was concentrated in vacuo and the remaining colorless syrup, containing
The fraction with R
f 0.05 (n-hexane/diethyl ether 1:2) afforded 107 mg (30%) of colorless amorphous
C36H30O11 (638.6): calcd. C 67.71, H 4.73; found C 67.56, H 4.62.
MS (FD, 20 mA): m/z = 638 (30 %, M+), 517 (100%, M+ − 121).
1H NMR (300 MHz, CDCl3) δ: 4.04 and 4.21 (each 1 H-m, glycol CH 2CH2OBz), 4.45 (dd, 1 H, 6 HA), 4.61 (m, 3 H, 6-HB, glycol CH 2OBz), 4.84 (ddd, 1 H, 5 H), 5.11 (s, 1 H, 1 H), 5.91 (dd, 1 H, 4 H), 6.17 (d, 1 H, 3 H), 7.2–8.2 (m, 20 H, 4 C6 H 5CO); J 3,4 = 10.2, J 4,5 = 10.2, J 5,6 = 5.0, J 6A,6B = 12.3 Hz.
13C NMR (75.5 MHz, CDCl3) δ: 62.4 (glycol CH2OBz), 63.4 (C-6), 66.8 (glycol CH2CH2OBz), 68.5 (C-5), 70.5 (C-4), 75.6 (C-3), 99.2 (C-1), 191.4 (C-2), 164.6, 165.1, 166.0, 166.3 (4 C6H5 CO).
Footnotes
Acknowledgments
The author thanks Prof Dr M Reggelin for the opportunity to work in his group and Dr V Schmidts for helpful discussions of the 1H and 13C NMR data.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.