
Abstract
The in vitro anti-inflammatory and skin moisturizing activities of Pilea martini (Levl.) Hand.-Mazz. were investigated on lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages and human immortalized keratinocytes. Chromatographic analysis was performed to identify the chemical composition of the extracts. Pilea martini extracts significantly suppressed LPS-induced nitric oxide, prostaglandin E2, interleukin 6, and tumor necrosis factor α production in dose-dependent manners. In addition, the extracts inhibited LPS-induced inducible nitric oxide synthase and cyclooxygenase-2 proteins and their mRNA expression through causing a downregulation of nuclear factor-κB, activator protein 1, and mitogen-activated protein kinase signaling cascades. The extracts increased the production of hyaluronic acid levels and enhanced the expression levels of both filaggrin and serine palmitoyltransferase regulation. Liquid chromatography-mass spectroscopy analysis showed that the extracts contained 6 different compounds (malic acid, tryptophan, chlorogenic acid, caffeic acid, p-coumaric acid, and isoquercetin) that may contribute to their bioactivities. Taken together, Pilea martini extract showed remarkable promise as an anti-inflammatory and moisturizing agent.
Pilea martini, family Urticales, is mainly found in tropical and subtropical regions of the world. Pilea has been traditionally used for various medicinal purposes in Vietnam. 1 P. martini extract, obtained from roots, stems, leaves, flowers, and berries, has been subjected to little research. In our study, we tried to demonstrate the potential anti-inflammatory effects of the extract.
Recently inflammation and its relationship to cancer is attracting much attention. 2 -4 In fact, inflammation of injured tissues in response to external insult leads to enhanced cell proliferation that can be linked to cancer. 5 -7 Inflammation has been known to play an important role in the development of cancer, as well as many human diseases. 8 Previous studies have shown that nitric oxide synthase (NOS) is related with cancer and inflammation. Nitric oxide (NO) can be stimulated by various cytokines, microbial products, immune complexes, and other activators. Activation of NOS increases the level of nitric oxide (NO), which induces inflammation. 9,10
Interaction between lipopolysaccharide (LPS) and Toll-like receptor 4 (TLR4) can be activated in pro-inflammatory cells. 11,12 This interaction causes the intracellular signaling of MyD88 with TLR4, which can induce the activation of nuclear factor-κB (NF-κB), activator protein 1 (AP-1) and mitogen-activated protein kinases (MAPKs) p38, ERK, and c-Jun NH2-terminal kinase (JNK). 13 The activation of these transcription factors expresses the inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF-α) . 14 Also, iNOS and COX-2 can regulate NO and prostaglandin E2 (PGE2) production. 15 Such enhanced production of PGE2, NO, IL-6, and TNF-α can lead to inflammatory responses. 16,17 However, overexpression of these inflammatory cytokines has high potential for various systemic disorders and damage, including death. 18,19 Previous studies have been focused on anti-inflammatory mechanisms through the suppression of NF-κB and MAPKs signaling pathway, including production of iNOS, COX-2, and various cytokines. 20 -23
Ceramides are a type of sphingolipid, which consist of a saturated fatty acid moiety and a sphingoid base. Ceramides can exist in the stratum corneum (SC), which plays an important role as a skin barrier with a water-holding function. 24 More than 12 types of ceramides are found in the SC; these are synthesized by several enzymes like serine palmitoyltransferase (SPT), glucosylceramide synthase, sphingomyelin synthase, ceramide synthase, β-glucocerebrosidase, and acid sphingomyelinase . 25 -31 According to a previous study, keratinocytes may respond to skin inflammation and pro-inflammatory cytokines by releasing several inflammatory mediators during the initiation of an inflammatory response. 32
In this study, we focused on the anti-inflammatory effects of Pilea martini in LPS-induced mouse macrophage RAW 264.7 cells and its possible skin moisturizing properties on human skin cells HaCaT. Therefore, we evaluated the production of NO and PGE2, and expression levels of various pro-inflammatory cytokines in RAW 264.7 cells. Furthermore, we evaluated the hyaluronic acid production and expression of SPT and filaggrin levels in HaCaT cells. Overall, we suggest that the blockage of NF-κB, AP-1, and MAPKs signaling may contribute to the anti-inflammatory and moisturizing properties of P. martini.
Results and Discussion
Pilea Extract Suppressed the Production of LPS-Induced Nitric Oxide in RAW 264.7 Cells
We examined the cytotoxicity of Pilea extract on RAW 264.7 cells. Cells were pretreated with the extract for 2 hours and additionally treated with LPS (1 µg/mL) for 24 hours before cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) assay. After Pilea extract and LPS treat, cells were incubated with MTT solution for 2 h, then incubated for MTT lysis for overnight at 37℃ in 5% CO2. As shown on results (Figure 1(A)), even concentration of extract was increasing, it exhibited low cytotoxicity on RAW 264.7 cells. It shows on LPS treated RAW 264.7 cells either. To investigate whether the extract had anti-inflammatory effects on nitrite production, RAW 264.7 cells were plated on a 6-well plate and treated as above. Supernatants were obtained from the Pilea extract-treated cells and mixed with Griess reagent then measured using a plate enzyme-linked immunosorbent assay (ELISA) reader. The results showed that nitric oxide was induced by LPS, but the Pilea extract significantly reduced the LPS-induced nitric oxide production (Figure 1(B)).

Inhibition of LPS-induced nitric oxide production by Pilea extract in RAW 264.7 cells. RAW 264.7 cells were seeded and pretreated with various indicated concentrations of Pilea extract for 2 hours and treated with LPS (1 µg/mL) for total 24 hours. (A) After 24-hour treatment, cell viability was analyzed by 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide assay. (B) Nitrite production in conditioned media was measured by Griess reaction assay. Standard concentrations of sodium nitrite were dissolved in fresh media and compared with each treatment group. Values represent the mean ± SD of triplicateexperiments (*** P < 0.001). (C) Pilea-treated cells were harvested and whole-cell lysates were prepared for western blot analysis. Same amount of proteins was prepared (10 µg/lane), separated on SDS-PAGE, and transferred to nitrocellulose membrane to probe iNOS and COX-2 by antibodies. (D) RAW 264.7 cells were seeded and pretreated with various indicated concentrations of Pilea extract for 2 hours and treated with LPS (1 µg/mL) for total 24 hours. Cells were harvested and RNA was extracted and examined for RT-PCR. iNOS and COX-2 were reverse transcribed and separated on 1% or 2% agarose gel. (E) Conditioned media from Pilea-treated cells was prepared, then amount of PGE2 was determined by PGE2 enzyme-linked immunosorbent assay kit. Values represent the mean ± SD of triplicateexperiments (** P < 0.005, *** P < 0.001). COX-2, cyclooxygenase-2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; iNOS, inducible nitric oxide synthase; LPS, ipopolysaccharide; PGE2, prostaglandin E2; RT-PCR, reverse transcription polymerase chain reaction; SDS-PAGE, sodium dodecyl-polyacrylamide gel electrophoresis.
Pilea Extract Had Suppression Effects on LPS-Induced iNOS and COX-2 Expression
Next, we examined the effects of Pilea extract on LPS-induced iNOS and COX-2 expression by western blot analysis. Cells were pretreated with various concentrations of Pilea extract for 2 hours and induced by LPS for 24 hours. Whole-cell lysates were obtained from the cells, and equal amount of proteins were prepared for western blot analysis. iNOS and COX-2 were induced by LPS, but Pilea extract clearly suppressed the expression of proteins (Figure 1(C)). Also, we examined the iNOS and COX-2 mRNA expression levels by reverse transcription polymerase chain reaction (RT-PCR). Cells were treated as above, then mRNA was extracted from whole cells and cDNA prepared for RT-PCR. The results showed that iNOS and COX-2 mRNA expression were reduced by Pilea extract treatments (Figure 1(D)).
LPS-Induced PGE2 Production Was Suppressed by Pilea Extract in RAW 264.7 Cells
Next we measured LPS-induced PGE2 production. RAW 264.7 cells were pretreated with Pilea extract for 2 hours and additionally treated with LPS for 24 hours. The supernatants from the extract-treated cells were prepared to measure PGE2 production using a PGE2 ELISA kit. LPS-induced PGE2 production was shown to be concentration-dependently suppressed by Pilea extract treatment (Figure 1(E)).
LPS-Induced Phosphorylation of P38, ERK, and JNK Was Inhibited by Pilea Extract
We next confirmed the effects of Pilea extract on LPS-induced phosphorylation of p38, ERK, and JNK. RAW 264.7 cells were pretreated with Pilea extract (50 µg/mL) for 2 hours and induced by LPS (1 µg/mL) for various time intervals. Equal amounts of whole-cell lysates were prepared for western blot analysis and expression of protein levels was observed using an enhanced chemiluminescence (ECL) kit. The results show that time-dependent LPS-induced phosphorylation of p38, ERK, and JNK expression was increased, but Pilea extract treatments clearly reduced the LPS-induced phosphorylation expression in RAW 264.7 cells. However, there was no significant change on p38, ERK, and JNK (Figure 2(A–C)). Specifically, phosphorylation of ERK increased up to 15 minutes after treatment with LPS and Pilea extract, after which it gradually decreased. However, there were no changes in p38, ERK, and JNK expression.

Suppressive effects of Pilea extract on LPS-induced mitogen-activated protein kinases activation in RAW 264.7 cells. RAW 264.7 cells were seeded and pretreated with 50 µg/ml of Pilea extracts for 2 hours and treated with LPS (1 µg/mL) for indicated time intervals. Phosphorylation of (A) p38, (B) ERK, and (C) JNK was probed by phospho-p38, p38, phospho-ERK, ERK, phospho-JNK, and JNK. All experiments were performed independently at least 3 times. JNK, c-Jun NH2-terminal kinase; LPS, lipopolysaccharide.
Pilea Extract Suppressed LPS-Induced NF-κB and AP-1 Activation in RAW 264.7 Cells
Because NF-κB and AP-1 can regulate the expression of iNOS and COX-2, we examined whether Pilea extract also had modulatory effects on NF-κB and AP-1 activation. RAW 264.7 cells were pretreated with Pilea extract for 2 hours and additionally treated with LPS for 24 hours. Then nuclear extracts were prepared from cells for electrophoretic mobility-shift assay (EMSA). Both LPS-induced NF-κB and AP-1 were clearly suppressed, concentration-dependently, by Pilea extract treatments (Figure 3(A) and (B)). Then RAW 264.7 cells were pretreated with Pilea extract for 2 hours and induced by LPS for various time intervals. The cytoplasmic and nuclear extracts were prepared for western blot analysis. As shown in Figure 3(C), Pilea extract blocked the LPS-induced degradation of IκB-α protein. In addition, nuclear translocation of LPS-induced phospho-p65, p65, c-Jun, and c-Fos was suppressed by the extract (Figure 3(D) and (E)).

Suppression effects of Pilea extract on LPS-induced NF-kB activation in RAW 264.7 cells. RAW 264.7 cells were pretreated with various indicated concentrations of Pilea extract for 2 hours, then exposed to LPS (1 µg/mL) for total 24 hours. Nuclear extracts from cells were prepared to evaluate DNA binding activation of (A) NF-κB and (B) AP-1 by electrophoretic mobility-shift assay. Oct-1 was used as loading control. (C) Cells were pretreated with 50 µg/mL of Pilea extract for 2 hours then stimulated by LPS (1 µg/mL) for indicated time intervals. Equal amounts of cytoplasmic extracts were prepared for western blot analysis, proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotansferred to nitrocellulose membranes. Proteins were probed for anti-IκB-α antibody and β-actin was used as loading control. (D and E) Cells were pretreated with Pilea extract (50 µg/mL) for 2 hours and simulated by LPS for indicated time intervals, cells were harvested, and nuclear extracts were prepared for western blot analysis. These proteins were probed by anti-phospho-p65, anti-p65, anti-c-Jun, and anti-c-Fos. Lamin B antibody was used as loading control. AP-1, activator protein 1; LPS, lipopolysaccharide; NF-κB, nuclear factor κB.
Pilea Extract Inhibited LPS-Induced Production of Pro-Inflammatory Cytokines IL-6 and TNF-α in RAW 264.7 Cells
We next measured the inhibitory effects of Pilea extract on the LPS-induced IL-6 and TNF-α production in RAW 264.7 cells. Cells were pretreated with Pilea extract for 2h, then incubated with LPS for total 24 h. Supernatants were obtained from the cells and then measured using a mouse ELISA kit. The experiment was conducted in accordance with the experimental method provided by the kit. The results showed that IL-6 and TNF-α production was induced by LPS, however, Pilea extract significantly reduced the production of both LPS-induced IL-6 and TNF-α (Figure 4(A) and (B)).
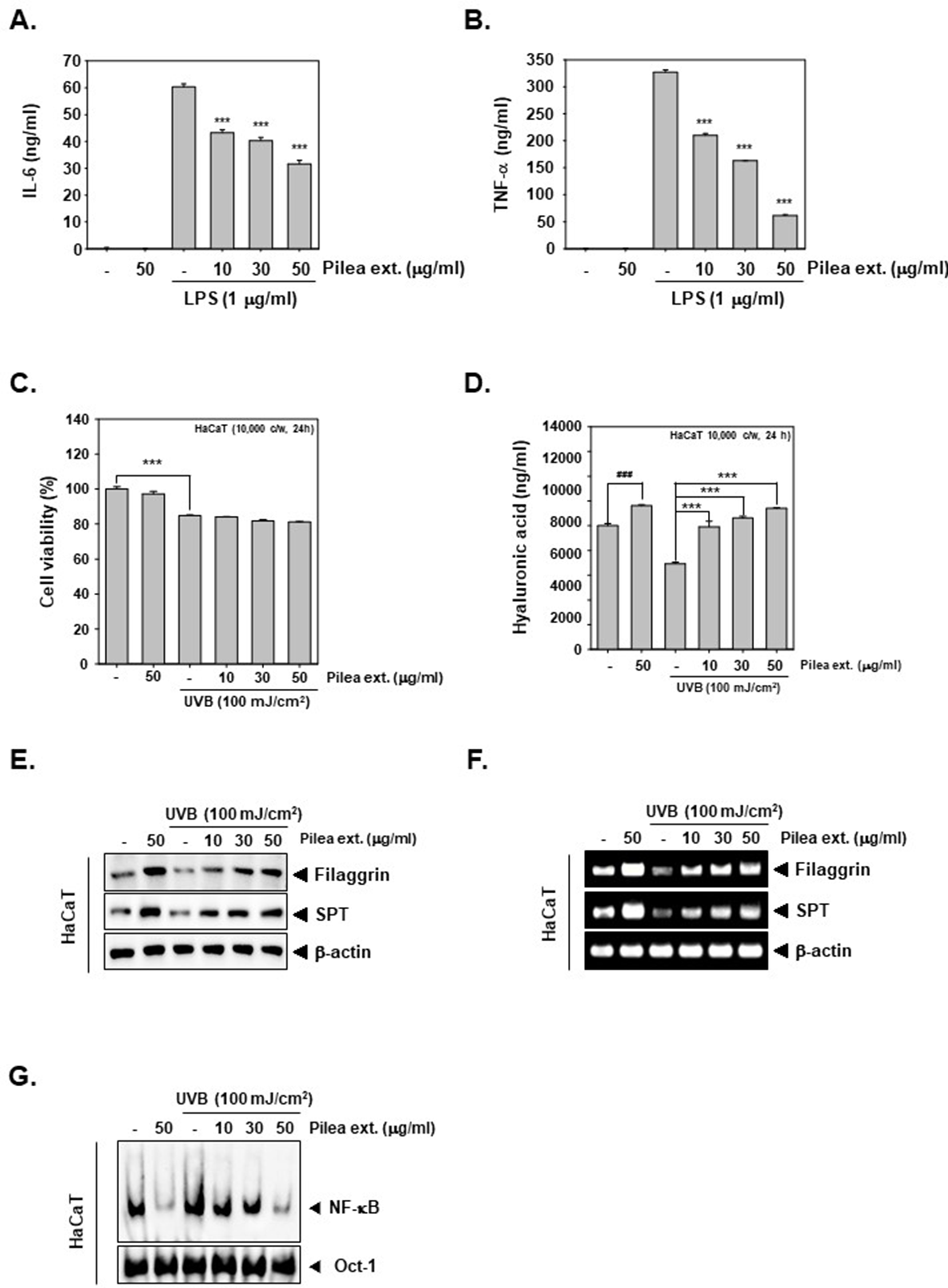
Suppressive effect of LPS-induced proinflammatory cytokines by Pilea extract in RAW 264.7 cells and moisturizing effects with increase of hyaluronic acid production by Pilea extract in HaCaT cells. RAW 264.7 cells were seeded and pretreated with various indicated concentrations of Pilea extract for 2 hours and then treated with LPS (1 µg/mL) for total 24 hours. Conditioned media were obtained from cells treated with each concentration and then (A) IL-6 and (B) TNF-α levels were measured by IL-6 and TNF-α ELISA kit. All experiments were performed at least 3 times individual repeats. Values represent the mean ± SD of triplicateexperiments (*** P < 0.001). (C) HaCaT cells were pretreated with Pilea extract and exposed by UVB for total 24 hours. Then cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide assay. Values represent the mean ± SD of triplicateexperiments (*** P < 0.001). (D) HaCaT cells were treated with concentration-dependent Pilea extract (0, 10, 30, and 50 µg/mL) for 24 hours. Supernatants were obtained from Pilea extract pretreated, and then exposed to UVB HaCaT cells and hyaluronic acid production were measured by hyaluronan acid ELISA kit. Values represent the mean ± SD of triplicateexperiments (*** P < 0.001, ### P < 0.001). (E) Expression levels of filaggrin and SPT were evaluated by western blot analysis. Proteins were probed by anti-filaggin, and anti-SPT antibodies. (F) mRNA expression levels of filaggrin and SPT were analyzed by reverse transcription polymerase chain reaction. (G) Nuclear factor-κB-DNA binding activity was analyzed upon nuclear extracts by electrophoretic mobility-shift assay. All experiments were performed independently at least 3 times. ELISA, enzyme-linked immunosorbent assay; IL-6, interleukin 6; LPS, lipopolysaccharide; SPT, serine palmitoyltransferase; TNF-α, tumor necrosis factor alpha.
Pilea Extract Increased the Production of Hyaluronic Acid in HaCaT Cells
We then confirmed the effects of Pilea extract on the production of hyaluronic acid in human skin HaCaT cells. First, we evaluated the cytotoxicity of Pilea extract against HaCaT cells. Cells were pretreated with Pilea extract for 2 hours, then exposed to UVB (100 mJ/cm2) and incubated for 24 hours. As shown in Figure 4(C), Pilea extract did not exhibit any substantial cytotoxicity; however, UVB exposure induced significant reduction of cell viability in HaCaT cells. Because hyaluronic acid plays a role in skin water retention, we measured its production in HaCaT cells after 2 hours of Pilea extract treatment, followed by exposure to UVB (100 mJ/cm2). 33 Figure 4(D) shows that hyaluronic acid production was increased, concentration dependently, by treatment with Pilea extract, thereby confirming the moisturizing effect of the extract in HaCaT cells.
Pilea Extract Induced the Expression of Filaggrin and SPT Levels in HaCaT Cells
We examined filaggrin and SPT expressions, which play an important role in the skin hydration process. HaCaT cells were treated with Pilea extract for 2 hours, followed by exposure to UVB (100 mJ/cm2). Then the whole-cell extracts were evaluated for filaggrin and SPT expression levels by western blot analysis. As shown in Figure 4(E), both filaggrin and SPT levels in HaCaT cells were increased in a concentration-dependent manner by treatment with Pilea extract. Next, we examined the mRNA expression levels of filaggrin and SPT by RT-PCR. As seen in Figure 4(F), UVB exposure decreased the filaggrin and SPT expressions in HaCaT cells, but both levels were increased, concentration-dependently, by treatment with Pilea extract.
Pilea Extract Suppressed the DNA Binding Activity of NF-ĸB in HaCaT Cells
Next we investigated the potential of Pilea extract to alter the DNA binding ability of NF-ĸB in HaCaT cells. Cells were treated with Pilea extract for 2 hours, followed by exposure to UVB (100 mJ/cm2). Thereafter, nuclear extracts were analyzed by EMSA. NF-κB-DNA binding activity was induced by UVB exposure, but Pilea extract, concentration-dependently, suppressed NF-κB activation in HaCaT cells (Figure 4(G)).
LC-MS Analysis for Identification of Chemical Components of Pilea Extract
We conducted a liquid chromatography-mass spectrometry (LC-MS) analysis of the extracts to determine their chemical profiles; many peaks were observed (Figure 5(A) and (B)). Details of the components corresponding to individual peaks are shown in Table 1. Identification of each component in the extracts was first analyzed by comparison with authentic standards with MasterView in PeakView software. Six compounds (malic acid, tryptophan, chlorogenic acid, caffeic acid, p-coumaric acid, and isoquercetin) were identified with the use of authentic standards. For further identification of the components, the LC-QTOF base peak chromatogram-collected information-dependent acquisition (IDA) scan mode was processed to search and screen components using exact masses and fragmentation ions against an in-house library and databases such as Chemspider, Metlin, and MS bank. The others were tentatively identified by matching with the in-house library and databases (Table 1).

Pilea extract induced the hyaluronic acid production and expression of both filaggrin and SPT in HaCaT cells. (A) Base peak chromatograms of the Pilea extract and extracted ion chromatograms of 6 authentic standards (B) analyzed by ultra-high-performance liquid chromatography-quadrupole time-of-flight in negative ion mode. (C) Targets of Pilea extract in HaCaT cells. COX-2, cyclooxygenase 2; IL-6, interleukin 6; iNOS, inducible nitric oxide synthase; MAPK, mitogen-activated protein kinase; PGE2, prostaglandin E2; SPT, serine palmitoyltransferase.
Identification of Chemical Components Authentically or Tentatively in Pilea Extract.

*1: Authentic standard.
*2: In-house MS/MS library (SCIEX).
*3: Metlin.
*4: MS bank.
Discussion
The effects of natural products from fruit, vegetables, and plants on various human diseases have been studied. 34 -38 However, there has been little study on the effects of Pilea martini extract on inflammatory diseases, and so we focused on this.
Our purpose was to investigate the anti-inflammatory effects of Pilea extract on RAW 264.7 mouse macrophage cells. Because the NF-κB and MAPK signaling pathways play an important role in inflammatory reactions, 39,40 we evaluated whether Pilea extract could suppress inflammation through downregulating the various pro-inflammatory transcription factors and gene products. 41 We demonstrated that the extract suppressed the phosphorylation of p38, ERK, and JNK, and translocation of NF-κB, c-Fos, and c-Jun in LPS-induced RAW 264.7 macrophage cells. Also, Pilea extract downregulated COX-2, iNOS, TNF-α, and IL-6. These results showed that the extract can demonstrate potent anti-inflammatory effects through downregulating NF-κB and MAPK signaling pathways in LPS-induced RAW 264.7 cells.
Our study showed that Pilea extract suppressed LPS-induced iNOS, COX-2, and PGE2 release in a concentration-dependent manner. Expression of the mRNA level of iNOS and COX-2 was suppressed by Pilea extract and thereafter, its protein levels were also clearly attenuated. MAPK signals play an important role in the inflammatory reaction, as well as NF-κB (PMID: 28086233;). 42,43 Pilea extract significantly downregulated the phosphorylation of MAPKs such as p38, ERK, and JNK in LPS-stimulated RAW 264.7 macrophage cells. Then AP-1, which is composed of c-Fos and c-Jun, can be activated and promote transcription. 44,45 Pilea extract downregulated AP-1 expression through the suppression of c-Jun and c-Fos in LPS-induced RAW 264.7 cells. In LPS-stimulated cells, IκB-α degradation can promote translocation of p65 activated into the nucleus. 46 -48 This study exhibited that Pilea extract suppressed NF-κB activation by causing downregulation of IκB-α degradation, as confirmed by EMSA and western blot analysis. Pro-cancer inflammatory mediators such as PGE2, IL-6, and TNF-α can be produced by complex interactions of various immune cells. 49,50 Interestingly, the production of these pro-cancer inflammatory mediators was clearly suppressed by Pilea extract through downregulation of the MAPKs signaling pathway and NF-κB activation in LPS-stimulated RAW 264.7 macrophage cells.
In general, moisturizing can maintain water-holding and skin barrier functions, which can contribute to maintain skin homoeostasis to prevent microorganisms or chemical invasion from the outside environment. Filaggrin is closely related to the SC as a structure protein, which is the most upper layer of skin and functions to protect water loss and prevent invasion into the skin from the outside environment and also controls epidermis differentiation. 51 -53 When filaggrin is degraded into amino acids, such as glutamine and histidine, these can transform into pyrrolidone carboxylic acid trans-urocanic, which can contribute to the generation of natural moisturizing factors to promote water-holding and water-loss prevention in skin through binding with water in the SC. 54 Ceramides de novo biosynthesis can be catalyzed by SPT to mediate a condensation reaction of L-serine with palmitoyl-CoA. Because of its active participation in these reactions, SPT has been considered a key enzyme to control the sphingolipid level in ceramide generation. 26
In conclusion, Pilea extract demonstrated potential anti-inflammatory effects by downregulation of the MAPKs signaling pathway and NF-κB/AP-1 activation in LPS-induced RAW 264.7 cells and promotion of the moisturizing properties in HaCaT cells. Moreover, the production of various inflammatory mediators such as PGE2, IL-6, and TNF-α was also suppressed by the extract, and finally activation of the inflammatory cascade was blocked. Furthermore, Pilea extract recovered the hyaluronic acid production and expression of both filaggrin and SPT following UVB exposure of HaCaT cells (Figure 5(C)). Our study provides convincing evidence in a cellular model and future in vivo studies can further establish the application of the anti-inflammatory and moisturizing effects of Pilea extract in different inflammatory ailments.
Materials and Methods
Reagents
Pilea martini (H. Lév.) Hand.-Mazz. extracts (Pilea extract) were obtained from the International Biological Material Research Center (Daejeon, Korea). The plant was collected in Xiangzhoucun, Changguling Forest Farm, Jinggangshan, Jiangxi Prvince, China in 2014 and authenticated by the curator of the Institute of Botany, at the Chinese Academy of Sciences (IBCAS), Xianchun Zhang. A voucher specimen recoded as “KRIB 0054093” was deposited in the herbarium of the Korea Research Institute of Bioscience and Biotechnology. The dried and refined whole plant (37 g) was extracted with 400 mL of 99.9% (v/v) methanol with repeating sonication (15 minutes) and resting (2 hours) for 3 days at 45 °C. The resultant product was filtered through non-fluorescence cottons, and concentrated in a rotary evaporator (N-1000SWD, EYELA) under reduced pressure at 45 °C. Finally, 2.52 g of methanol extract (Pilea extract) was obtained by freeze-drying. This was stored as a 100 µg/mL stock solution in dimethyl sulfoxide at −20 ℃, and diluted in the culture media for in vitro experiments. MTT, Tris base, glycine, NaCl, sodium dodecylsulfate (SDS), and bovine serum albumin were purchased from Sigma-Aldrich (St Louis, MO USA); LightShift Chemiluminescent EMSA kit from Thermo Fisher Scientific Inc.; mouse IL-6, TNF-α,and hyaluronan ELISA kit from R&D Systems (Minneapolis, MN, USA); PGE2 ELISA kit from Thermo Fisher Scientific (Waltham, MA, USA); anti-mouse iNOS and anti-mouse COX-2 antibodies from BD Biosciences (Becton-Dickinson, Franklin Lakes, NJ, USA); anti-IκBα, anti-phospho-p65, anti-p65, anti-c-Jun, anti-c-Fos, anti-β−actin, anti-filaggrin, anti-SPT, and anti-Lamin B from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and anti-phospho p38, anti-p38, anti-phospho-ERK, anti-ERK, anti-phospho-JNK, and anti-JNK from Cell Signaling Technology (Beverly, MA, USA).
Cell Line and Culture Condition
Mouse RAW 264.7 macrophage cells and human HaCaT keratinocyte cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The RAW 264.7 cells were cultured in RPMI-1640 medium containing 10% inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin. The HaCaT cells were cultured in Dulbecco’s modified Eagle low-glucose medium containing 10% inactivated FBS and 1% penicillin-streptomycin. Cells were cultured and maintained at 37 ℃ in 5% CO2.
MTT Assay
To evaluate the cytotoxicity of the Pilea extract using the MTT assay, RAW 264.7 cells (1 × 104 cells/well) were seeded on a 96-well plate and incubated overnight in 37 °C. Cells were pretreated with the Pilea extract (50 µg/mL) for 2 hours and then treated with LPS (1 µg/mL) for 24 hours. After treatment, 30 µL of MTT solution (5 mg/mL) was added for 2 hours and MTT lysis buffer for at least 8 hours. The lysed MTT formazans were measured by VARIOSKAN LUX (Thermo Fisher Scientific Inc, Waltham, MA, USA) at 570 nm.
Western Blot Analysis
The Pilea extract and either LPS-treated or nontreated cells were harvested and whole-cell extracts, cytoplasmic extracts, and nuclear extracts were isolated from the cells to prepare for sodium dodecyl-polyacrylamide gel electrophoresis (SDS-PAGE) gel evaluation. Equal amounts of proteins were prepared and resolved by SDS-PAGE, then electrotransferred to a nitrocellulose membrane. Membranes were blocked with 5% skimmed milk for 2 hours at room temperature and probed with target primary antibodies overnight at 4 ℃. The membranes were then incubated with horse radish peroxidase (HRP) at room temperature for 1 hour. For detection, an ECL kit (EZ-Western Lumi Femto, DOZEN) was used.
RT-PCR for RNA Analysis
RAW 264.7 cells were pretreated with various concentrations of Pilea extract and LPS for 24 hours. Cells were harvested, suspended in trizol, then incubated with chloroform and isopropanol. Extracted RNA was reverse transcribed into cDNA for RT-PCR, which was performed with iNOS and COX-2 at 94 ℃ for 5 minutes, 94 ℃ for 30 seconds, 58 ℃ for 30 seconds, and 72 ℃ for 30 seconds, with 30 cycles, and an extension at 72 ℃ for 7 minutes. RT-PCR for Filaggrin and SPT was performed by incubating cDNA at 94 ℃ for 5 minutes, 94 ℃ for 30 seconds, 57 ℃ for 30 seconds, and 72 ℃ for 30 seconds, with 30 cycles, followed by an extension at 72 ℃ for 5 minutes. Glyceraldehyde-3-phosphate dehydrogenase was used as control and all experiments were performed at least 3 times.
Electrophoretic Mobility Shift Assay
Pilea extract and LPS-treated RAW 264.7 cells were prepared to isolate nuclear extracts. NF-κB and AP-1 DNA binding activation was evaluated using NF-κB oligonucleotide (5′‐AGTTGAGGGGACTTTCCCAGGC‐3′ and 5′‐GCCTGGAAAGTCCCCTCAACT‐3′), 5′‐biotinylated AP-1 oligonucleotide (5′‐CGCTTGATGAGTCAGCCGGAA-3′ and 5′‐TTCCGGCTGACTCATCAAGCG-3′). Oct-1 (5′-TTCTAGTGATTTGCATTCGACA-3′ and 5′-TGTCGAATGCAAATCACTAGAA-3′) was used as loading control.
Nitrite Assay
RAW 264.7 cells (1 × 104 cells/well) were plated in 6-well plates. Cells were pretreated with Pilea extract (0, 10, 30, and 50 µg/mL) for 2 hours, and induced with LPS (1 µg/mL) for total 24 hours. Nitrate assay was examined with Griess reagent in cultured medium. Equal volumes of culture medium were mixed with Griess reagent and calibrated by sodium nitrite solution standard (Sigma). Then absorbance was measured using a plate ELISA reader at 540 nm.
Measurement of PGE2 Release in RAW 264.7 Cells
To measure PGE2 production from RAW 264.7 cells, cells (5 × 105 cells/well) were plated on a 6-well plate. Cells were pretreated with Pilea extract (0, 10, 30, and 50 µg/mL) for 2 hours, and induced with LPS (1 µg/mL) for 24 hours. The cultured medium from the Pilea extract-treated cells was harvested and PGE2 measured by a PGE2 ELISA Kit.
Measurement of IL-6 and TNF-α Production in RAW 264.7 Cells
To confirm the anti-inflammatory effects of Pilea extract, the levels of pro-inflammatory cytokines such as IL-6 and TNF-α were measured. RAW 264.7 cells (5 × 105 cells/well) were plated on a 6-well plate, pretreated with with Pilea extract (0, 10, 30, and 50 µg/mL) for 2 hours, and induced with LPS (1 µg/mL) for 24 hours. Supernatants from the Pilea extract treated cells were measured using a mouse ELISA kit.
Measurement of Hyaluronic Acid Production in HaCaT Cells
HaCaT cells (5 × 105 cells/well) were plated on a 6-well plate and treated with different concentrations of Pilea extract (0, 10, 30, and 50 µg/mL) for 24 hours. Supernatants were obtained from each sample, then hyaluronic acid production was measured using a hyaluronan ELISA kit.
UVB Irradiation
HaCaT cells were seeded in a 6-well plate (5 × 105 cells/well). The cells were pre-treated with Pilea extract for 2 hours, then exposed to 100 mJ/cm2 of UVB radiation using a CL-1000 Ultraviolet Crosslinker (Ultra-violet products Ltd, Cambridge, UK).
Qualitative Analysis of Pilea Extracts by LC-MS
Approximately 10 mg of extract was shaken with 1 mL of 70% ethanol using a vortex mixer for 30 seconds. The supernatants were filtered through a 0.2-µm polytetrafluoroethylene syringe filter (Thermo Scientific). Finally, the filtrate was transferred to an LC sample vial before use. The LC-MS system consisted of a Thermo Scientific Vanquish UHPLC system (Thermo Fisher Scientific, Sunnyvale, CA, USA) with a Shim-pack GIS-ODS (3.0 mm × 100 mm, 3 µm; Shimadzu) and a Triple TOF5600 +mass spectrometer system (Triple TOF MS; QTOF, SCIEX, Foster City, CA, USA). The QTOF MS, equipped with a Duospray ion source, was used to complete the high-resolution experiment. The LC gradient used a mobile phase A containing 0.1% formic acid in water and a mobile phase B containing 0.1% formic acid in acetonitrile. The flow rate was kept constant at 0.8 mL/min and the injection volume was 2 µL. The gradient elution system began at 5% B for 0.8 minutes, 5%-20% B from 0.8 to 2.5 minutes, 20%-32% B from 2.5 to 8.5 minutes, 32%-70% from 8.5 to 11.5 minutes, then increased to 100% B at 12.0 minutes, held at 100% B for 3 minutes, and then returned to the initial conditions for re-equilibration. Mass data acquisition was performed with a Triple TOF 5600+ in negative ion mode using the following parameters: source temperature set at 450 ℃ with a curtain gas flow of 25 L/min (GS1 and GS2 both 50), the ion spray voltage was set at −4500 V, declustering potential was 30 V, and the collision energy was 10 V. High-purity nitrogen gas was used for the nebulizer/Duospray and curtain gases. The QTOF and IDA scan was operated with a mass range of 80‐1200 m/z. Precursor and product ion calibration were performed in both high-sensitivity and high-resolution modes using a calibrant delivery system prior to analysis. Data acquisition and processing for qualitative analysis were carried out using Analyst TF 1.7, PeakVeiw2.2 and MasterView (SCIEX, Foster City, CA, USA).
Statistical Analysis
All numerical values are represented as the mean ± SD. Statistical significance of the data compared with the untreated control was determined using the Student’s unpaired t‐test. Significance was set at *P <0.05, **P <0.01, and ***P <0.001.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the National Research Foundation of Korea funded by the Korean government (NRF-2017M3A9E4057932).