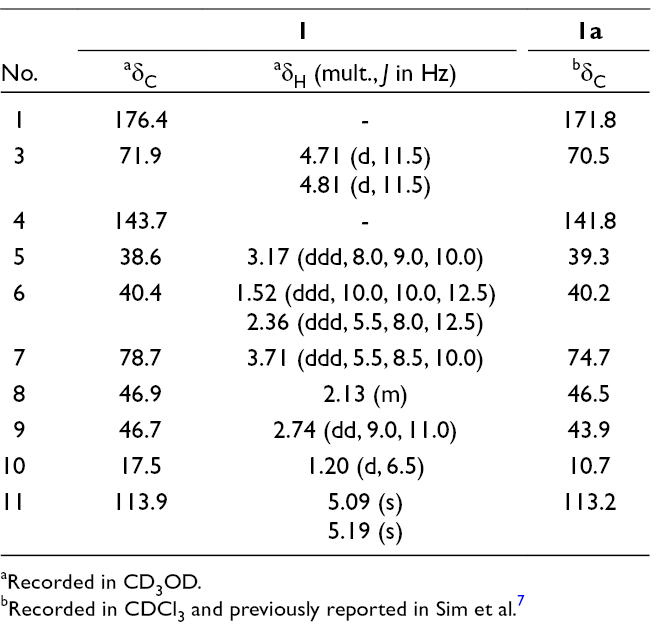
Three new constituents (1-3) were isolated from the whole plant of Balanophora laxiflora. Their chemical structures were elucidated by extensive analysis of high-resolution electrospray ionization mass spectrometry and nuclear magnetic resonance spectroscopy. Absolute configurations of new compounds were determined by circular dichroism spectra and modified Mosher’s method. The isolated compounds weakly inhibited on both NO production and COX-2 mRNA expression in RAW264.7 macrophages.
Balanophora laxiflora (Balanophoraceae) is a parasitic plant and usually parasitizes at the roots of evergreen trees. It is widely distributed in the tropical or subtropical forests of Vietnam, Laos, Cambodia, and China.1,2 The whole plant of B. laxiflora is used in the folk medicinal remedies for antidote, anti-inflammation, improvement of blood circulation, and as a tonic drug.2,3 Literature survey indicates that B. laxiflora and other Balanophora species are rich sources of phenolic compounds such as lignans, chalcones, hydrolysable tannins, and acyl glucose of phenolic acids.2-6 In the present study, the whole plant of B. laxiflora was subjected to chemical investigation and led to the isolation of three new compounds including an iridoid (1), a rare natural occurring 1-hydroxy-1,3-diarylpropan-2-one glucoside (2), and an aryltetralin lignan glucoside (3). The anti-inflammatory effects of those compounds were evaluated by inhibition of NO production and reducing COX-2 mRNA expression in RAW264.7 cells.
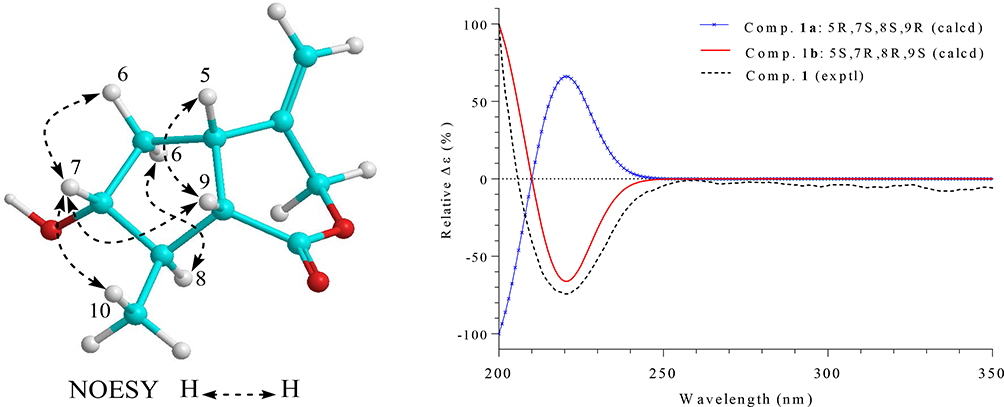
Compound 1 was obtained as a white amorphous powder. Its molecular formula was determined to be C10H14O3 by a quasi-molecular ion peak at m/z 183.1016 [M+H]+ (calcd. for C10H15O3, 183.1021) in the high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) and in conjunction with the 13C-NMR data. The infrared (IR) spectrum of 1 showed stretching vibration bands at 3427 and 1711 cm-1, suggesting the presence of hydroxyl and carbonyl functional groups, respectively. The 1H-NMR spectrum of 1 contained signals of an exo-olefinic methylene (δH 5.09 and 5.19), an oxygenated methylene (δH 4.71 and 4.81), an oxygenated methine (δH 3.71), and a doublet methyl group (δH 1.20). The 13C-NMR spectrum of 1 revealed of 10 carbon signals, which were further classified by heteronuclear single quantum coherence spectra as two nonprotonated carbons, four methines, three methylenes, and one methyl carbon. A deshielded carbon signal at δC 176.4 was assigned to a carbonyl group. Two olefinic carbon signals at δC 113.9 and 143.7 were demonstrated for the presence of a C-C double bond. In the 1H-1H COSY spectra, a 5-membered ring structure was established by the correlations of neighboring protons including H-5 (δH 3.17)/ H2-6 (δH 1.52, 2.36)/ H-7 (δH 3.71)/ H-8 (δH 2.13)/ H-9 (δH 2.74)/ H-5. On the other hand, a COSY correlation between H-8 and methyl group (H3-10, δH 1.20) indicated the attachment of methyl group at C-8 (Figure S1, Supplementary data). The HMBC correlations between H3-10 (δH 1.20) and C-9 (δC 46.7)/ C-8 (δC 46.9)/ C-7 (δC 78.7), and chemical shift of methine C-7 (δH 3.71, δC 78.7) suggested the presence of hydroxy group at C-7. Moreover, HMBC correlations between H-8 (δH 2.13)/ H-9 (δH 2.74) and deshielded carbon (δC 176.4) indicated that C-1 was a carbonyl carbon (Figure S1, Supplementary data). Location of exocyclic double bond at C-4/C-11 was confirmed by HMBC correlations between H2-11 (δH 5.09, 5.19) and C-5 (δC 38.6)/ C-4 (δC 143.7)/ C-3 (δC 71.9). Finally, signal of oxygenated methylene C-3, HMBC correlation between H2-3 (δH 4.71, 4.81) and carbonyl carbon C-1 (δC 176.4) indicated δ-lactone linkage between C-3 and C-1. Thus, the planar structure of 1 was established and similarly with a previously synthesized compound (1a).7 However, significant difference in their NMR data (Table 1) suggested that they are stereoisomers and expected difference at chiral centers C-7 and/or C-8. Relative configuration of 1 was examined by nuclear Overhauser effect spectroscopy (NOESY) analysis. A NOESY correlation between H-5 (δH 3.17) and H-9 (δH 2.74) indicated a cis-fused 5/6-bicyclic system (Figure 1). Therefore, H-5 and H-9 were proposed to be β-orientation as notably reported for the iridoids.8-11 The NOESY correlations between H-7 (δH 3.71) and H-9 (δH 2.74)/ H3-10 (δH 1.20)/ Hβ-6 (δH 2.36), Hα-6 (δH 1.52) and H-8 (δH 2.13) suggested for the β-orientation of C-10 and α-orientation of OH-7. Experimental and Time dependent density functional theory calculated circular dichroism (CD) spectra of 1 observed negative Cotton effect at 220 nm, indicating absolute configurations of 5S,7R,8R,9S (Figure 1). Consequently, compound 1 was determined to be a new iridoid and named as balanolaxin (Figure 2).
bRecorded in CDCl3 and previously reported in Sim et al.7
Important nuclear Overhauser effect spectroscopy correlations, experimental CD, and theoretical calculated CD spectra of possible enantiomers (1b and 1c) of compound 1.
Chemical structures of compounds 1, 2, and 3.
Compound 2 was isolated as a pale-yellow amorphous powder. A quasi-molecular ion peak at m/z 459.1274 [M+Na]+ in the HR-ESI-MS and 13C-NMR data indicated molecular formula of 2 to be C21H24O10 (calcd. for C21H24O10Na, 459.1267). The 1H-NMR spectrum of 2 displayed characteristic signals for an ABX-coupled aromatic proton system [δH 6.50 (1H, br d, J = 7.8 Hz), 6.75 (1H, br s), 7.07 (1H, br d, J = 7.8 Hz)] and an ′BB′-coupled aromatic proton system [δH 6.66 (2H, d, J = 7.8 Hz), 6.88 (2H, d, J = 7.8 Hz)], indicating the presence of a 1,2,4-trisubstituted and a 1,4-disubstituted benzene ring, respectively. The 13C-NMR and heteronuclear single quantum coherence spectra of 2 contained signals for 21 carbons including 6 nonprotonated carbons, 13 methines, and 2 methylenes. Of these, 6 carbinol signals at δC 103.5, 74.9, 78.2, 71.7, 77.9, and 62.8 were typically recognized for a glucopyranosyl group. Twelve aromatic carbons (δC 104.9–160.6) were assigned for 2 benzene rings. Remaining 3 carbons at δC 210, 76.6, and 44.9 belonged to a ketone, an oxygenated methine, and an sp3 methylene group, respectively. The HMBC correlations between H-1 (δH 5.32) and C-2 (δC 210.0)/ C-1′ (δC 120.6)/ C-2′ (δC 157.9)/ C-6′ (δC 132.1), H2-3 (δH 3.61, 3.56) and C-2/C-1″ (δC 126.4)/ C-2″ (δC 131.7)/ C-6″ (δC 131.7) indicated a 1,3-diaryl propan-2-one structure, specifically, a para-disubstituted benzene ring at C-3 and 1,2,4-trisubstituted benzene ring at C-1 (Figure S1, Supplementary data). Additional hydroxy group at C-1 was confirmed by its oxygenated methine signals δH 5.32/ δC 76.6. The large coupling constant of H-6′/H-5′ (J = 7.8 Hz, ortho-coupled protons), and HMBC correlations between H-6′ (δH 7.07) and C-2′ (δC 157.9)/ C-4′ (δC 160.6) supported for the assignment of 2 sp2 oxygenated carbons C-4′ and C-6′. Another sp2 oxygenated carbon C-4″ was deduced by HMBC correlation between H-2″ (δH 6.88)/ H-6″ (δH 6.88) and C-4″ (δC 157.2). The HMBC correlations between H-1 (δH 5.32)/ H-6′ (δH 7.07)/anomeric proton H-1‴ (δH 4.87) and C-2′ (δC 157.9) indicated glucopyranosyl group at C-2′. Acid hydrolysis of 2 gave sugar and aglycone portions. The presence of d-glucose in the sugar portions was confirmed by high-performance liquid chromatography (HPLC) analysis of its thiocarbamoylthiazolidine derivative in comparison with those of authentic samples.12 Aglycone portion was reacted with S and R-MTPA-Cl reagents to give R-MTPA and S-MTPA esters, respectively.13 The difference in the 1H-NMR data between S-MTPA and R-MTPA esters indicated the S-configuration of C-1 (Figure S2, Supplementary data). Additionally, the S-configuration of C-1 was also agreed with a negative Cotton effect at 246 nm (Δε: −4.14) and a positive Cotton effect at 299 nm (Δε: +7.81) in the CD spectrum of 2 as described in the literature.14 Consequently, structure of compound 2 was established and named as balanophoroside A (Figure 2).
Compound 3 was isolated as a pale-yellow amorphous powder. Its molecular formula was deduced as C26H34O11 by a quasi-molecular ion peak at m/z 545.1998 in the HR-ESI-MS (calcd. for C26H34O11Na, 545.1999). The 1H-NMR spectrum of 3 contained signals for one AX-coupled aromatic proton [δH 6.82 and 6.74 (each 1H, s)], one ABX-coupled aromatic proton [δH 6.71 (1H, d, J = 2.0 Hz), 6.67 (1H, d, J = 8.0 Hz), 6.46 (1H, dd, J = 2.0, 8.0 Hz)], one anomeric proton [δH 4.68 (1H, d, J = 7.5 Hz)], and two methoxy groups [δH 3.87 and 3.78 (each 3H, s)]. The 13C-NMR spectrum of 3 contained signals for 26 carbons. Of these, 6 carbinol signals at δC 102.8, 74.8, 77.8, 71.2, 78.1, and 62.4 were typically recognized for a glucopyranosyl group. Two carbon signals at δC 56.8 and 56.4 were assigned for 2 methoxy groups. Remaining 18 carbon signals including 12 aromatic carbons (δC 113.4−149.3), 2 hydroxymethylene carbons (δC 65.5 and 63.4), 3 methines (δC 35.4, 44.6, and 46.7), and 1 methylene (δC 33.1) suggested for an aryltetralin lignan backbone, containing 2 hydroxymethylene groups (C-6a and C-7a). The HMBC correlations between H-1 (δH 6.74) and C-8 (δC 46.7)/ C-3 (δC 149.3), methoxy protons (δH 3.87), and C-3 indicated location of a methoxy group at C-3. Meanwhile, HMBC correlations between H-4 (δH 6.82) and C-5 (δC 33.1)/ C-2 (δC 146.3), anomeric proton (δH 4.68) and C-2 indicated an O-glucopyranosyl group at C-2 (Figure S1, Supplementary data). In the 1,3,4-trisubstituted benzene ring, HMBC correlations between H-2′ (δH 6.71)/ H-6′ (δH 6.46) and C-4′ (δC 145.9), H-5′ (δH 6.67)/methoxy protons (δH 3.78) and C-3′ (δC 148.3) suggested for the presence of a hydroxy group at C-4′, a methoxy group at C-3′, respectively. The presence of d-glucose in compound 3 was also confirmed by acid hydrolysis and HPLC analysis of thiocarbamoyl-thiazolidine derivatives (Supplementary data). On the other hand, the close similarity NMR spectral data between 3 and sargentodoside A (3a)15 (Table 2) suggested that the aglycone moiety of 3 and 3a are a pair of enantiomers. Because of containing an aryltetralin chromophore, absolute configurations at chiral carbons C-6, C-7, and C-8 of compound 3 were elucidated by CD analysis, which has been reviewed in many reports.16 The CD spectrum of 3 observed Cotton effects (Δε: +9.46 (238 nm), +3.32 (275 nm), −1.72 (292 nm)) which were in opposite trend with 3a ([θ]: −50018 (237 nm), −21053 (274 nm), +5901 (291 nm) for 6S,7R,8R configurations),15 indicating 6R,7S,8S configurations. Thus, compound 3 was determined to be a new aryltetralin lignan glycoside and named as balanophoroside B (Figure 2).
1H and 13C NMR Data for Compounds 3 and 3a in CD3OD.
The anti-inflammatory activity of compounds 1-3 was evaluated by inhibition of the NO production17 and reducing COX-2 mRNA expression18 in lipopolysaccharide-stimulated RAW264.7 cells. As shown in Figure 3, the compounds showed weak inhibition in both NO production and COX-2 mRNA expression in the activated RAW264.7 cells at a tested concentration of 10 µM. Their inhibitory values were in the range from 9.91% to 35.61%. Among the three compounds, compound 2 displayed the best NO inhibition (22.39%); meanwhile, compound 1 inhibited higher in the COX-2 mRNA expression (35.61%). At a concentration of 10 µM, all of the compounds (1-3) did not show significant cytotoxic effect against RAW264.7 cells by MTT assay.
Effect of compounds 1-3 (10 µM) on the NO production and COX-2 expression in LPS-stimulated RAW264.7 cells, P < 0.05 vs #vehicle or *LPS groups.
Experimental
General
Optical rotation was measured on a Jasco P2000 polarimeter. Circular dichroism spectra were acquired on a Chirascan spectrometer. NMR spectra were recorded on Bruker 500 MHz or 600 MHz spectrometer using Tetramethylsilane as an internal standard. High-resolution electrospray ionization mass spectrometry was acquired on an Agilent 6530 Accurate Mass Q-TOF LC/MS system.
Plant Material
The whole plants of B. laxiflora Hemsley were collected at Sapa, Lao Cai province, Vietnam in November 2015 and taxonomically determined by botanist Nguyen The Cuong, Institute of Ecology and Biological Resources, VAST. A voucher specimen (NCCT-P01) was deposited at the Institute of Marine Biochemistry, VAST.
Extraction and Isolation
The dried powdered whole plant of B. laxiflora (4.0 kg) was extracted with MeOH in ultrasonic bath for 3 times (each 6 L, 30 minutes, at 40°C). After removal of solvent in vacuo, 300 g of crude extract was suspended in 2 L distilled water and separated in turn with dichloromethane and ethyl acetate. Ethyl acetate soluble fraction (110 g) was fractionated on a silica gel column, eluting with dichloromethane/methanol (40/1, 20/1, 10/1, 5/1, 3/1, 1/1, v/v, each 1 L) to give 6 fractions E1-E6. Fraction E3 (9.0 g) was repeatedly chromatographed on a silica gel column, eluting with dichloromethane/acetone (5/1, v/v) to yield 5 fractions E3A-E3E. Fraction E3C was first purified on a reversephase (RP-18) CC, eluting with methanol/water (3/1, v/v) and then futher purified by preparative TLC (dichloromethane/ methanol, 7/1, v/v) to give compound 1 (4 mg). Fraction E5 was subjected on a silica gel column chromatography and eluted with acetone/dichloromethane/water (2/1/0.1, v/v/v) to give 4 fractions E5A-E5D. Fraction E5B was chromatographed using silica gel column and eluent of dichloromethane/ methanol/water (6/1/0.1, v/v/v) to give 3 fractions E5B1-E5B3. Fraction E5B1 was purified on an RP-18 column, eluting with acetone/water (1/2, v/v) to give compound 2 (14 mg). Compound 3 (8 mg) was isolated from fraction E5B3 using RP-18 column and acetone/water (2/5, v/v) as eluent.
Balanolaxin (1)
White amorphous powder.
: +19.3 (c 0.1, MeOH).
CD (MeOD) Δε (λ nm): −3.31 (220).
IR (KBr): 3427, 2974, 2921, 1711, 1615, 1482, 1393 cm-1.
HR-ESI-MS: m/z 545.1998 [M+ Na]+ (calcd. for C26H34O11Na, 545.1999).
Theoretical Calculation of CD Spectra for Compound 1
Conformational searches were performed by Spartan 14 program. Conformations were optimized and subjected to TDDFT calculation using Gaussian 09 program. The calculated CD spectra were combined on the Boltzmann distribution of each stable conformer using SpecDis v1.64 software. Two possible enantiomers of 1 (1b: 5R,7S,8S,9R and 1c: 5S,7R,8R,9S) were submitted to conformational searches at ground state with semi-empirical AM1 set. The initial stable conformers with Boltzmann distributions over 1.0% (8 conformers) were optimized by DFT calculations at the B3LYP/6-31G(d,p) basic set and polarizable continuum model (PCM) calculation of the solvent methanol. The optimized conformers were then subjected to TDDFT calculations at the B3LYP/6-31G(d,p) level in the presence of methanol as a PCM. The CD spectra at 30 excited states for each conformer were collected and summed to obtain theoretical CD spectra of each enantiomer. Half-band was taken as ζ = 0.3 eV. After UV corrections, the calculated spectra of enantiomers were shifted by 3 nm.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2015.75.
Supplemental Material
References
1.
KanisA.CuongVV.VidalJE.HansenB.CussetC. Flora of Cambodia, Laos, and Vietnam. Vol 14. French, Paris: National Museum of Natural History; 1973:49-57.
2.
SheG-M.ZhangY-J.YangC-R. Phenolic constituents from Balanophora laxiflora with DPPH radical-scavenging activity. Chem Biodivers. 2009;6(6):875-880.doi:10.1002/cbdv.200800139
3.
QuangDN.SoTC.ThanhNTPet al. Balanochalcone, a new chalcone from Balanophora laxiflora Hemsl. Nat Prod Res. 2018;32(7):767-772.doi:10.1080/14786419.2017.1359172
4.
JiangZH.HiroseY.IwataH.SakamotoS.TanakaT.KounoI. Caffeoyl, coumaroyl, galloyl, and hexahydroxydiphenoyl glucoses from Balanophora japonica. Chem Pharm Bull. 2001;49(7):887-892.doi:10.1248/cpb.49.887
5.
HoS-T.TungY-T.ChengK-C.WuJ-H. Screening, determination and quantification of major antioxidants from Balanophora laxiflora flowers. Food Chem. 2010;122(3):584-588.
6.
TanakaT.UeharaR.NishidaK.KounoI. Galloyl, caffeoyl and hexahydroxydiphenoyl esters of dihydrochalcone glucosides from Balanophora tobiracola. Phytochemistry. 2005;66(6):675-681.doi:10.1016/j.phytochem.2004.10.018
7.
SimJ.YoonI.YunH.AnH.SuhY-G. Divergent synthetic route to new cyclopenta[c]pyran iridoids: syntheses of jatamanin A, F, G and J, gastrolactone and nepetalactone. Org Biomol Chem. 2016;14(4):1244-1251.doi:10.1039/C5OB02147B
8.
DindaB.DebnathS.HarigayaY. Naturally occurring iridoids. A review, part 1. Chem Pharm Bull. 2007;55(2):159-222.doi:10.1248/cpb.55.159
9.
DindaB.DebnathS.HarigayaY. Naturally occurring secoiridoids and bioactivity of naturally occurring iridoids and secoiridoids. A review, part 2. Chem Pharm Bull. 2007;55(5):689-728.doi:10.1248/cpb.55.689
10.
DindaB.ChowdhuryDR.MohantaBC. Naturally occurring iridoids, secoiridoids and their bioactivity. An updated review, part 3. Chem Pharm Bull. 2009;57(8):765-796.doi:10.1248/cpb.57.765
11.
DindaB.DebnathS.BanikR. Naturally occurring iridoids and secoiridoids. An updated review, part 4. Chem Pharm Bull. 2011;59(7):803-833.doi:10.1248/cpb.59.803
12.
TanakaT.NakashimaT.UedaT.TomiiK.KounoI. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem Pharm Bull. 2007;55(6):899-901.doi:10.1248/cpb.55.899
13.
OhtaniI.KusumiT.KashmanY.KakisawaH. High-field FT NMR application of Mosher's method. The absolute configurations of marine terpenoids. J Am Chem Soc. 1991;113(11):4092-4096.doi:10.1021/ja00011a006
14.
TanakaH.SudoM.KawamuraTet al. Antibacterial constituents from the roots of Erythrina herbacea against methicillin-resistant Staphylococcus aureus. Planta Med. 2010;76(9):916-919.doi:10.1055/s-0029-1240849
15.
ZengX.WangH.GongZet al. Antimicrobial and cytotoxic phenolics and phenolic glycosides from Sargentodoxa cuneata. Fitoterapia. 2015;101:153-161.doi:10.1016/j.fitote.2015.01.008
16.
KurtánT.AntusS.PescitelliG. Electronic CD of benzene and other aromatic chromophores for determination of absolute configuration. In Comprehensive chiroptical spectroscopy. Wiley and Sons; 2012:73-114.
17.
Van KiemP.CuongLCV.TrangDTet al. New alkaloids and anti-inflammatory constituents from the leaves of Antidesma ghaesembilla. Nat Prod Commun. 2017;12(1):11-14.
18.
JinZ.YangY-Z.ChenJ-X.TangY-Z. Inhibition of pro-inflammatory mediators in RAW264.7 cells by 7-hydroxyflavone and 7,8-dihydroxyflavone. J Pharm Pharmacol. 2017;69(7):865-874.doi:10.1111/jphp.12714
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.