
Abstract
Background
Melanoma represents one of the most aggressive skin cancers, responsible for over 75% of skin cancer-related deaths despite comprising only 5% of cases. Despite therapeutic advances, patient responses remain variable and unpredicv. The Signaling Lymphocytic Activation Molecule (SLAM) family regulates immune cell communication, with SLAMF8 being predominantly expressed on myeloid cells. However, SLAMF8's specific role in melanoma pathogenesis remains largely unexplored.
Methods
In this retrospective integrative study, we systematically investigated SLAMF8's role in melanoma through multi-omics analyses using TIMER, GEPIA, and UALCAN databases, following the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines for observational data reporting. Functional studies were conducted in A-375 and SK-MEL-28 melanoma cell lines using siRNA-mediated knockdown, followed by migration, invasion, and proliferation assays. DNA methylation patterns were analyzed via the SMART database, while mutation profiles were examined using cBioPortal and COSMIC. Immune infiltration analysis was performed through TIMER, and pathway associations were investigated using Gene Set Enrichment Analysis and protein-protein interaction networks. Finally, two-sample Mendelian Randomization analysis assessed the causal relationship between SLAMF8 expression and melanoma susceptibility.
Results
SLAMF8 expression was significantly higher in metastatic melanoma compared to primary tumors, with expression patterns varying across disease stages and between sexes. Higher SLAMF8 expression correlated with improved disease-free and overall survival. Functional studies demonstrated that SLAMF8 knockdown significantly enhanced melanoma cell proliferation, migration, and invasion. DNA methylation analysis revealed significant negative correlations between methylation at specific CpG sites and SLAMF8 expression, with hypermethylation associated with worse survival outcomes. Mutation analysis identified alterations in 10.21% of melanoma patients. Immune infiltration studies demonstrated strong correlations between SLAMF8 expression and enhanced immune cell presence. GSEA linked SLAMF8 to critical immune pathways including allograft rejection, inflammatory response, and interferon signaling. Mendelian Randomization analysis established a protective causal relationship between SLAMF8 and melanoma risk (OR = 0.39, 95% CI = 0.21–0.74, p = 3.34e-03).
Conclusion
Our study demonstrates that SLAMF8 plays a critical role in melanoma by suppressing tumor progression and modulating the immune microenvironment. Elevated SLAMF8 expression in metastatic melanoma is associated with improved patient survival, suggesting its utility as a prognostic biomarker. Furthermore, its tumor-suppressive effects and immune-regulatory functions highlight SLAMF8 as a promising therapeutic target for melanoma treatment strategies.
Introduction
Melanoma represents one of the most aggressive forms of skin cancer, arising from malignant transformation of melanocytes and characterized by its rapid progression, metastatic potential, and high mortality when diagnosed at advanced stages. 1 While melanoma accounts for approximately 5% of all skin cancers, it is responsible for over 75% of skin cancer-related deaths worldwide. 2 Skin cutaneous melanoma (SKCM), the most prevalent subtype, exhibits remarkable heterogeneity in its molecular landscape, clinical presentation, and treatment response profiles. 3 Despite significant advances in therapeutic options, including targeted therapies against BRAF/MEK mutations and immune checkpoint inhibitors (ICIs) targeting CTLA-4, PD-1, and PD-L1, clinical responses remain variable and unpredictable. 4 Approximately 40–60% of patients with advanced melanoma respond to ICI monotherapy, while combination immunotherapy may yield response rates of up to 60%, despite increased toxicity.5,6
The Signaling Lymphocytic Activation Molecule (SLAM) family plays a crucial role in immune cell communication and modulation. 7 Members of this family are differentially expressed on various immune cell types and contribute to both adaptive and innate immune responses. Recent studies have highlighted the involvement of SLAM family proteins in tumor immunology, particularly in shaping the tumor microenvironment (TME). For instance, SLAMF7 has been shown to drive T-cell exhaustion within the TME, thereby limiting effective anti-tumor immunity. 8 Additionally, SLAMF5 has been implicated in immunosuppressive mechanisms by regulating PD-1/PD-L1 expression in hematologic malignancies such as chronic lymphocytic leukemia and multiple myeloma. 9 In solid tumors, the upregulation of SLAMF4 in tumor-infiltrating CD8+ T cells of head and neck squamous cell carcinoma has been correlated with increased PD-1 and PD-L1 expression, further contributing to an immunosuppressive niche.10,11
SLAMF8 (also known as BLAME/CD353), a member of the SLAM family, mainly expressed on macrophages, neutrophils and dendritic cells, also on B cells. 12 Unlike other SLAM family proteins, which have been extensively studied in hematologic and solid tumors, the role of SLAMF8 in cancer remains largely unexplored. Initial findings have demonstrated its involvement in inflammatory signaling and bacterial phagocytosis, as well as its ability to influence macrophage polarization and dendritic cell migration. 13 In hematologic malignancies, SLAMF8 is expressed in and enhances the growth of anaplastic large cell lymphoma (ALCL) cells via SHP-2 activation, and it interacts with activated SHP-2 and ALK proteins in ALK-positive ALCL cells. 14 In glioma, high SLAMF8 expression is linked to poor prognosis, chemotherapy resistance, and enhanced immunosuppression, particularly in the mesenchymal subtype characterized by active immune and inflammatory responses. 15 Recent evidence suggests that SLAMF8 is associated with the prognosis of colorectal cancer and may serve as a potential novel immune checkpoint molecule. 16 Moreover, elevated SLAMF8 expression has been linked to improved responses to anti-PD-1 immunotherapy in gastrointestinal cancers, 17 which is particularly relevant given that PD-1 blockade is a standard and highly effective therapy for advanced melanoma. However, its specific role in melanoma has yet to be elucidated.
To address this, we conducted a retrospective observational study based on multi-omics databases, complemented by in vitro functional assays and Mendelian randomization analysis, to comprehensively investigate the role of SLAMF8 in melanoma progression and prognosis. By evaluating the correlation between SLAMF8 expression and melanoma progression, we aim to elucidate its clinical significance and mechanistic involvement in tumor development. This research seeks to deepen our understanding of melanoma biology and identify potential therapeutic targets to improve patient outcomes.
Methods
Study design
This study was designed as a retrospective observational study integrating public multi-omics datasets and in vitro experimental validation. The observational component adhered to the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines. Potential biases include sample heterogeneity across datasets, batch effects, and limited clinical annotation in public databases. In addition, Mendelian randomization analysis relies on core assumptions such as the absence of horizontal pleiotropy, which may not always hold. The retrospective nature of the study and reliance on existing data also limit the ability to infer definitive causality. These limitations were acknowledged and addressed where possible by applying rigorous statistical methods and complementary experimental validation.
Exploration of SLAMF8 expression in melanoma
To comprehensively investigate the expression profile of SLAMF8 in melanoma and its clinical relevance, we employed multiple bioinformatics platforms including TIMER, GEPIA, and UALCAN databases. The TIMER (Tumor IMmune Estimation Resource) 18 and GEPIA (Gene Expression Profiling Interactive Analysis) databases 19 were utilized to systematically explore SLAMF8 expression patterns across diverse tumor types from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) datasets, with particular emphasis on skin cutaneous melanoma (SKCM). Differential expression analysis between tumor and normal tissue samples was conducted to identify SKCM-specific alterations in SLAMF8 expression levels. Additionally, we performed survival analyses using these platforms to evaluate the potential prognostic significance of SLAMF8 in melanoma patients, including overall survival and disease-free survival assessments. To further characterize the clinical relevance of SLAMF8 in melanoma, we employed the UALCAN database 20 to investigate associations between SLAMF8 expression and various clinicopathological parameters. The observational component of this study complies with the STROBE guidelines.
Cell culture and reagents
From the National Collection of Authenticated Cell Cultures (Shanghai, China), we acquired two human melanoma cell lines: A-375 and SK-MEL-28. A-375 cells were maintained in DMEM medium while SK-MEL-28 cells were cultivated in 1640 medium (both from Keygene, China, Thermofisher). Both culture media were supplemented with 10% fetal bovine serum (Every Green) and 1% Penicillin-Streptomycin-Gentamicin Solution (Beyotime). Cell cultivation occurred at 37°C in a humidified environment with 5% CO2.
RNA interference
For SLAMF8 silencing, specific siRNAs were procured from Ribobio (Guangzhou, China), with sequence details provided in
Supplementary Table 1
Migration evaluation
A wound healing (scratch) assay was performed to assess cell migration. Cells were seeded in 6-well plates at a density of 1.5 × 105 cells/well and cultured until reaching full confluence. Experimental groups included the negative control (NC) group, si1-SLAMF8 group, and si2-SLAMF8 group. A sterile 100 μL pipette tip was used to create five parallel scratch wounds in each well. Subsequently, 1 mL of fresh culture medium was added to each well. Untreated cells served as the NC group. After 24 h of incubation, images of wound closure were captured using an inverted optical microscope. The migration area was quantified using ImageJ software, and cell migration was calculated as: Migration = [(initial wound area − wound area at 24 h) / initial wound area] × 100%.
Invasion analysis
Cell invasion was evaluated using Matrigel-coated Transwell chambers (8 μm pore size). A total of 2 × 104 cells were seeded into the upper chamber in 200 μL serum-free medium, while the lower chamber was filled with complete culture medium serving as a chemoattractant. Untreated cells were used as the negative control (NC) group. After 24 h of incubation at 37°C, non-invading cells on the upper surface of the membrane were gently removed using a cotton swab. The invaded cells on the lower surface were fixed with 4% formaldehyde, stained with 1% crystal violet, and imaged under an inverted microscope. The invasion rate was calculated as: Invasion rate = Number of cell migrations in the experimental group / Number of cell migrations in the control group.
Protein expression analysis
For protein extraction, cells were collected via scraping into SDS buffer containing protease and phosphatase inhibitor cocktails (Beyotime). Standard western blotting protocols were followed to analyze the protein levels of PCNA (D3H8P, 1:500) with β-actin (66009–1-Ig, 1:50,000) serving as loading control.
RT-PCR analysis of SLAMF8 expression
Total RNA was extracted from A-375 and SK-MEL-28 cells using the TRIzol reagent (Invitrogen, USA) according to the manufacturer's instructions. RNA concentration and purity were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). First-strand cDNA was synthesized from 1 μg of total RNA using the Maxima H Minus Reverse Transcriptase Kit (Thermo Fisher Scientific, USA) following the supplier's protocol. RT-PCR was performed using gene-specific primers for SLAMF8 ( Supplementary Table 1 ) and GAPDH as an internal control. PCR cycling conditions were as follows: 95 °C for 10 min, 35 cycles of 95 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. Relative SLAMF8 expression was quantified using the 2^-ΔΔCt method. Each experiment was performed in triplicate.
MTT (3-(4,5)-dimethylthiahiazo (-z-y1)-3,5- di- phenytetrazoliumromide) assay
Cells in the logarithmic growth phase were seeded into 96-well plates at a density of (5 × 10³ – 1 × 104 cells/well), optimized based on preliminary experiments. The experimental groups included the negative control (NC) group, si1-SLAMF8 group, and si2-SLAMF8 group. After incubation for 24 h at 37°C to allow adherence, the medium was replaced with fresh medium containing the test compounds (control wells received compound-free medium). Cells were then incubated for predetermined time points (e.g., 24, 48, and 72 h). Subsequently, 10–20 µL of MTT solution (5 mg/mL in PBS, final concentration 0.5 mg/mL) was added to each well, followed by incubation at 37°C for 4 h to allow formazan crystal formation. The supernatant was carefully aspirated, and 150 µL of DMSO or 10% SDS in 0.01 M HCl was added to dissolve the formazan. The plates were agitated for 10 min, and the absorbance was measured at 490 nm using a microplate reader. Cell viability was calculated using the formula: Cell viability (%) = (OD_sample / OD_control) × 100.
SLAMF8 methylation analysis in SKCM
To investigate the epigenetic regulation of SLAMF8 in melanoma, we performed comprehensive methylation profiling using the SMART (Survival analysis, Methylation Analysis, And Regulatory factor prediction Tool) database. 21 Initially, we conducted an overview analysis of methylation sites across the SLAMF8 gene. Next, we performed correlation analyses between methylation levels at individual CpG sites and SLAMF8 mRNA expression in SKCM samples from the TCGA dataset. Pearson correlation coefficients were calculated to quantify the strength and direction of these associations. Furthermore, we conducted survival analyses to evaluate the prognostic significance of SLAMF8 methylation patterns in SKCM patients. Kaplan-Meier analyses were performed to assess the relationship between methylation status at key regulatory sites and patient outcomes, including overall survival and disease-free survival.
SLAMF8 mutation analysis in melanoma
To characterize the mutational profile of SLAMF8 in melanoma, we utilized two well-established genomic databases: cBioPortal 22 and COSMIC 23 (Catalogue Of Somatic Mutations In Cancer). Through cBioPortal, we analyzed the TCGA-SKCM dataset to determine the frequency and types of SLAMF8 genetic alterations. The oncoprint visualization tool was employed to illustrate mutation patterns across melanoma patients. We further validated these findings using the COSMIC database, which provided additional information on SLAMF8.
Immune infiltration analysis of SLAMF8 in melanoma
To investigate the relationship between SLAMF8 and the tumor immune microenvironment in melanoma, we conducted comprehensive immune infiltration analyses using the TIMER database. Initially, we examined the association between SLAMF8 copy number alterations and infiltration levels of various immune cell populations in SKCM samples. We further explored the correlation between SLAMF8 expression levels and the abundance of different immune cell types, including B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells. Correlation coefficients were calculated while controlling for tumor purity to ensure robust assessment of immune infiltration patterns. Additionally, we conducted survival analyses to evaluate the prognostic significance of immune cell infiltration in relation to SLAMF8 expression.
Gene set enrichment analysis (GSEA)
To explore the functional pathways associated with SLAMF8 in melanoma, we performed Gene Set Enrichment Analysis (GSEA) 24 using transcriptomic data. The analysis was conducted with the hallmark gene sets from the Molecular Signatures Database (MSigDB) to identify significantly enriched biological pathways. Enrichment results were ranked based on normalized enrichment scores (NES) and false discovery rate (FDR) values, with FDR < 0.05 considered statistically significant.
Protein-Protein interaction (PPI) network construction
To further investigate the molecular interactions of SLAMF8, we constructed a protein-protein interaction (PPI) network using the STRING database (Search Tool for the Retrieval of Interacting Genes/Proteins). 25 The interaction confidence score was set at ≥ 0.4 (medium confidence) to include experimentally validated and computationally predicted interactions.
Functional enrichment analysis
Genes identified from the PPI network were subjected to Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analysis using Enrichr. 26 KEGG pathway analysis was used to determine the biological pathways associated with SLAMF8-interacting genes, while GO analysis was performed to classify these genes based on biological processes (BP), molecular functions (MF), and cellular components (CC). Pathways and terms with adjusted p-values < 0.05 were considered significantly enriched.
Mendelian randomization analysis
To assess the potential causal relationship between SLAMF8 expression and melanoma susceptibility, we performed a two-sample Mendelian Randomization (MR) analysis using the TwoSampleMR and MR-PRESSO packages in R. Instrumental variable (IV) selection: Genetic instruments for SLAMF8 expression were derived from cis-eQTLs (±300 kb) obtained from the GTEx v8 dataset via the OpenGWAS platform (ID: eqtl-a-ENSG00000158714). Only variants with genome-wide significance (p < 5 × 10−6) were retained. To reduce linkage disequilibrium, we applied LD clumping (r² < 0.3, clump window = 300 kb), retaining independent SNPs. Palindromic SNPs with intermediate allele frequencies were excluded during harmonization. Instrument strength was assessed by calculating F-statistics for each SNP (F=β2/SE2), and all included IVs had F > 10. Summary-level GWAS data for melanoma were obtained from the FinnGen consortium (finn-b-C3_MELANOMA_SKIN_EXALLC). Causal effects were primarily estimated using the inverse-variance weighted (IVW) method. Sensitivity analyses included MR-Egger regression, weighted median, and leave-one-out analysis. To identify and correct for potential horizontal pleiotropy and outlier SNPs, we additionally performed MR-PRESSO global and outlier tests (1000 simulations, p < 0.05). All MR estimates were reported as odds ratios (ORs) with 95% confidence intervals (CIs), and statistical significance was defined as p < 0.05.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8.0 (San Diego, CA, USA). For sample size estimation, a statistical power of 0.90 and a significance level (α) of 0.05 were applied, and the corresponding minimum sample size was determined based on the effect size derived from preliminary experiments. Data are expressed as mean ± standard deviation (SD). For comparisons between two groups, an unpaired two-tailed Student's t-test was used. For multiple group comparisons, two-way analysis of variance (ANOVA) was applied. All experiments were performed in triplicate biological replicates, with each group measured in at least three technical replicates. A p-value < 0.05 was considered statistically significant.
Results
Aberrant expression of SLAMF8 in melanoma and other cancers
We first analyzed the expression patterns of SLAMF8 across multiple cancer types and observed significant dysregulation in several malignancies. Notably, in melanoma, SLAMF8 expression was significantly higher in metastatic tumors compared to primary tumors and was also upregulated in tumor tissues compared to adjacent normal tissues (Figure 1A-C). Further stratification by cancer stage revealed a nonlinear expression pattern of SLAMF8. Stage 1 tumors exhibited higher SLAMF8 levels compared to Stage 2, followed by a significant increase in Stage 3 compared to Stage 2. This suggests that SLAMF8 may play distinct roles at different stages of melanoma progression, with potential involvement in both early tumor development and advanced disease progression (Figure 1D). Sex-based analysis indicated that SLAMF8 expression was significantly higher in female patients compared to males (p < 0.05) (Figure 1E). However, no significant differences were observed in SLAMF8 expression across different races, age groups, or body weight categories (p > 0.05). Survival analysis demonstrated that higher SLAMF8 expression correlated with better prognosis. In both disease-free survival (DFS) and overall survival (OS) analyses, patients with high SLAMF8 expression exhibited significantly prolonged survival compared to those with low expression levels (DFS: Logrank p = 0.02; HR = 0.57, p = 0.02; OS: Logrank p = 2.70e−05; HR = 0.57, p = 3.40e−05), indicating that SLAMF8 may serve as a favorable prognostic biomarker in melanoma (Figure 1F-G).
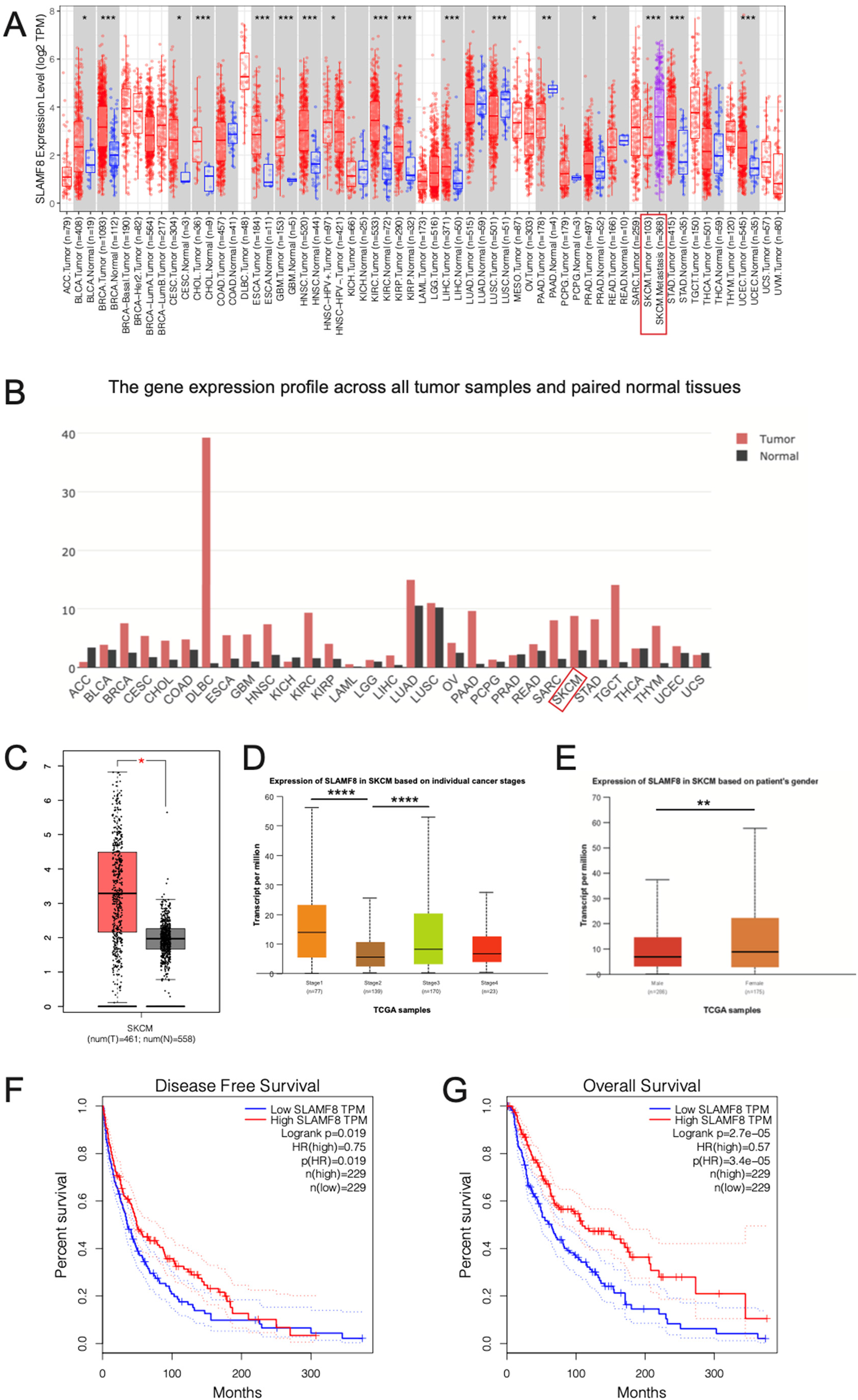
Aberrant expression of SLAMF8 in melanoma and other cancers. (A) Pan-cancer analysis of SLAMF8 expression across various malignancies reveals significant dysregulation in multiple cancer types. (* p < 0.05; ** p < 0.01; *** p < 0.001, Student's t-test). (B) SLAMF8 expression is significantly upregulated in melanoma tumor tissues compared to adjacent normal tissues. (C) Metastatic melanoma exhibits higher SLAMF8 expression than primary melanoma. (* p < 0.05, Student's t-test). (D) SLAMF8 expression shows a nonlinear pattern across melanoma stages, with elevated levels in Stage 1, a decrease in Stage 2, and a marked increase in Stage 3. (**** p < 0.0001, Student's t-test). (E) SLAMF8 expression is significantly higher in female patients compared to males.(** p < 0.01, Student's t-test). (F–G) Kaplan–Meier survival analyses demonstrate that higher SLAMF8 expression is associated with prolonged disease-free survival (DFS) and overall survival (OS) in melanoma patients. (DFS: Logrank p = 0.02; HR = 0.57, p = 0.02; OS: Logrank p = 2.70e−05; HR = 0.57, p = 3.40e−05).
SLAMF8 suppresses melanoma cell proliferation, migration, and invasion in vitro
To investigate the functional role of SLAMF8 in melanoma, we performed a series of in vitro assays using A-375 and SK-MEL-28 cell lines. RT-PCR analysis first confirmed that SLAMF8 expression was elevated in both melanoma cell lines compared with normal skin tissue (Supplementary Figure 1A). To further investigate its role, SLAMF8 expression was silenced using two independent siRNAs. Knockdown efficiency was verified by RT-PCR in both A-375 and SK-MEL-28 cells, which showed a marked reduction in SLAMF8 transcript levels following transfection (Supplementary Figure 1B-C). Wound healing assays demonstrated that the migratory ability of A-375 cells was significantly enhanced following SLAMF8 knockdown (Figure 2A-B, NC: 42.13 ± 3.56%, si1-SLAMF8: 74.52 ± 2.99%, si2-SLAMF8: 78.87 ± 1.14%, p < 0.0001), a trend that was also observed in SK-MEL-28 cells (Figure 2C-D, NC: 51.23 ± 2.69%, si1-SLAMF8: 71.18 ± 2.06%, si2-SLAMF8: 77.54 ± 3.26%, p < 0.0001). Similarly, Transwell invasion assays indicated that loss of SLAMF8 led to a marked increase in the invasive potential of A-375 cells (Figure 2E-F, NC: 46.19 ± 1.22%, si1-SLAMF8: 77.92 ± 0.79%, si2-SLAMF8: 78.89 ± 0.63%, p < 0.0001), with SK-MEL-28 cells showing comparable results (Figure 2G-H, NC: 47.75 ± 1.36%, si1-SLAMF8: 85.62 ± 1.69%, si2-SLAMF8: 87.02 ± 3.01%, p < 0.0001). Western blot analysis showed a notable upregulation of PCNA, a key marker associated with cell proliferation, upon SLAMF8 depletion, which was in line with the RT-PCR results (Figure 2I-J, A-375: NC 0.49 ± 0.02, si1-SLAMF8 1.12 ± 0.06, si2-SLAMF8 2.09 ± 0.08; SK-MEL-28: NC 0.46 ± 0.03, si1-SLAMF8 1.04 ± 0.02, si2-SLAMF8 1.54 ± 0.08; p < 0.0001). Moreover, MTT assays confirmed that SLAMF8 knockdown significantly promoted melanoma cell proliferation at 24, 48, and 72 h (Figure 2K-L).
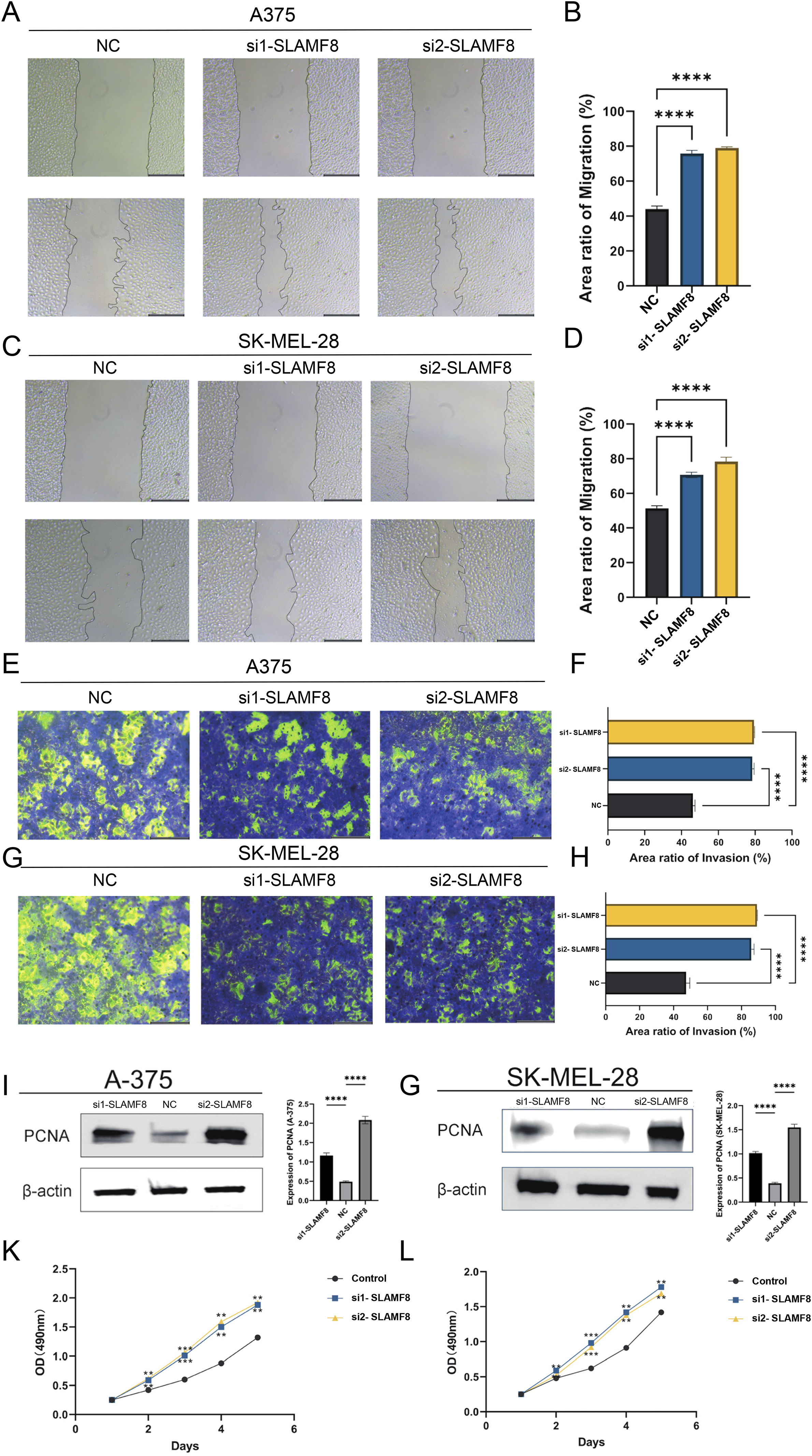
SLAMF8 suppresses melanoma cell proliferation, migration, and invasion in vitro. (A–D) Quantitative wound healing assays of A-375 and SK-MEL-28 cells after SLAMF8 knockdown. Cell migration was quantified as [(initial wound area − wound area at 24 h) / initial wound area] × 100% using ImageJ. Bar graphs represent mean ± SD of three independent experiments (n = 3), each performed in triplicate wells. (**** p < 0.0001, Student's t-test). (E–H) Transwell invasion assays indicate increased invasion following SLAMF8 silencing. Invasion rate = (number of migrated cells in experimental group / number of migrated cells in control group) × 100%. Data are shown as mean ± SD (n = 3 independent experiments). (**** p < 0.0001, Student's t-test). (I–J) Western blot analysis of PCNA protein expression, which is a key marker associated with cell proliferation, normalized to β-actin. Band intensities were quantified relative to negative control (NC) group and presented as mean ± SD (n = 3 independent experiments). (**** p < 0.0001, Student's t-test). (K–L) MTT assays measuring metabolic activity of A-375 and SK-MEL-28 cells after SLAMF8 knockdown. Optical density (O.D.) at 490 nm reflects relative cell viability. Data are presented as mean ± SD (n = 3 independent experiments, each with three technical replicates). (** p < 0.01; *** p < 0.001, two-way ANOVA).
SLAMF8 methylation and its association with gene expression and survival in melanoma
To further explore the epigenetic regulation of SLAMF8, we analyzed the DNA methylation status of seven CpG sites (cg07590721, cg10179949, cg17972058, cg04275881, cg07625783, cg06764092, and cg18084791) (Figure 3A). Correlation analysis between methylation levels and SLAMF8 expression revealed that five CpG sites (cg04275881, cg07625783, cg06764092, cg10179949, and cg18084791) exhibited a significant negative correlation with SLAMF8 gene expression (Figure 3B, p < 0.05), suggesting a potential regulatory role of DNA methylation in modulating SLAMF8 transcription. Next, we conducted survival analysis to evaluate the prognostic relevance of SLAMF8 methylation in melanoma patients (Figure 3C). The results indicated that higher methylation levels at cg07625783 (HR = 1.67, 95%CI = 1.28–2.18, p = 2e−04), cg06764092 (HR = 1.44, 95%CI = 1.10–1.88, p = 7.3e−03), and cg18084791 (HR = 1.45, 95%CI = 1.11–1.90, p = 5.5e−03) were significantly associated with worse overall survival. These findings suggest that hypermethylation at these sites may contribute to tumor progression and poorer clinical outcomes in melanoma.
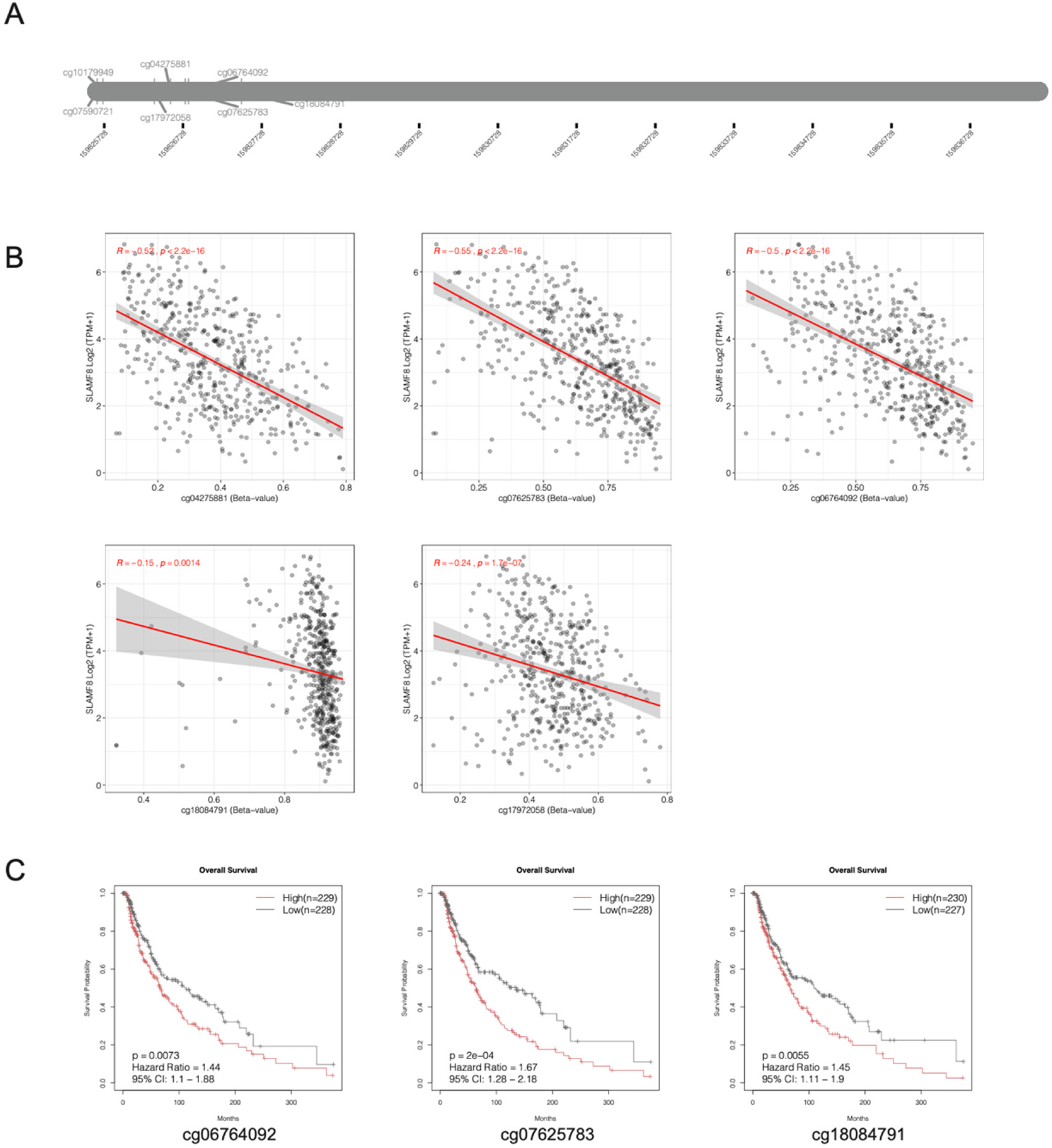
SLAMF8 methylation and its association with gene expression and survival in melanoma. (A) DNA methylation levels of seven CpG sites within the SLAMF8 gene (cg07590721, cg10179949, cg17972058, cg04275881, cg07625783, cg06764092, and cg18084791) were analyzed in melanoma samples. (B) Correlation analysis shows that five CpG sites (cg04275881, cg07625783, cg06764092, cg10179949, and cg18084791) are significantly negatively correlated with SLAMF8 mRNA expression levels (p < 0.05). (C) Kaplan–Meier survival analysis reveals that higher methylation at cg07625783 (HR = 1.67, 95%CI = 1.28–2.18, p = 2e−04), cg06764092 (HR = 1.44, 95%CI = 1.10–1.88, p = 7.3e−03), and cg18084791 (HR = 1.45, 95%CI = 1.11–1.90, p = 5.5e−03) is associated with poorer overall survival in melanoma patients, indicating a potential prognostic value of SLAMF8 methylation status.
Mutation Status of SLAMF8 in melanoma
The mutational landscape of SLAMF8 was assessed using data from cBioPortal and COSMIC databases to understand its potential role in melanoma. As shown in Figure 4A-B, SLAMF8 was found to be altered in 10.21% of 470 melanoma patients. Among these alterations, 3.62% (17 patients) exhibited mutations(mostly Missense Mutation), 2.98% (14 patients) showed amplifications, and 3.62% (17 patients) had high mRNA expression levels. Further analysis of COSMIC mutation data (Figure 4C) revealed a predominance of missense substitutions, which accounted for 42 mutations (61.76%). Additionally, there were 17 synonymous substitutions (25.00%), 4 other alterations (5.88%), and 2 nonsense substitutions (2.94%). The most common mutation types were G > A (49.12%) and C > T (38.60%), followed by A > G (7.02%) and G > T (5.26%)(Figure 4D).
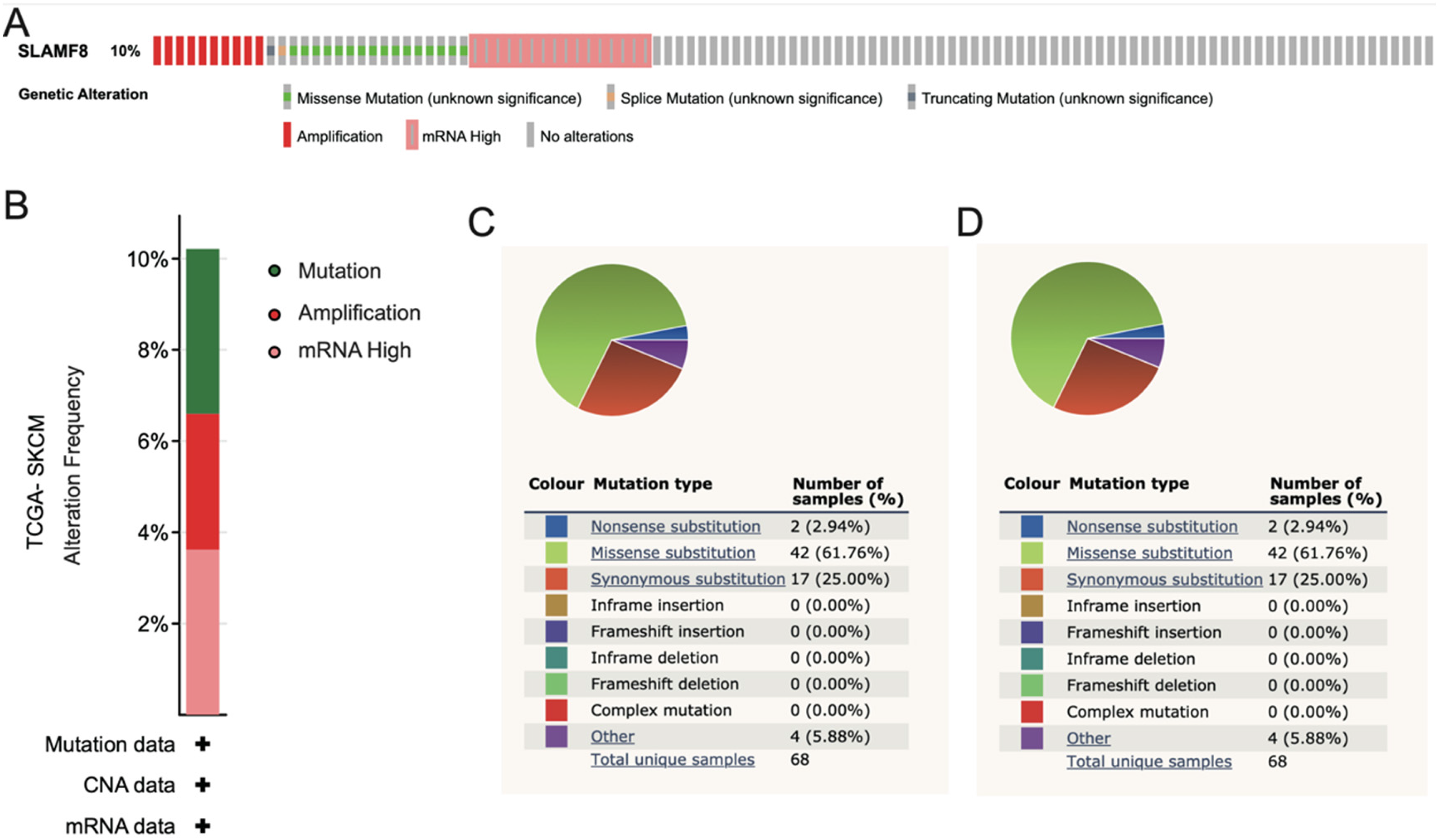
Mutation Status of SLAMF8 in melanoma. (A-B) Genomic alterations of SLAMF8 in melanoma were analyzed using cBioPortal data. SLAMF8 was altered in 10.21% of 470 melanoma patients, including mutations(mostly Missense Mutation) (3.62%), gene amplifications (2.98%), and high mRNA expression (3.62%). (C-D) Mutation data from the COSMIC database show that the majority of SLAMF8 mutations are missense substitutions (61.76%), followed by synonymous substitutions (25.00%), other alterations (5.88%), and nonsense substitutions (2.94%). The most frequent base substitution types were G > A (49.12%) and C > T (38.60%).
Tumor immune infiltration and its correlation with SLAMF8 in melanoma
In Figure 5A, we observed a strong correlation between SLAMF8 somatic copy number alterations (SCNAs) and immune cell infiltration levels in melanoma. Alterations in SLAMF8 were associated with significant changes in immune infiltration, highlighting its role in shaping the tumor immune microenvironment. Figure 5B shows that SLAMF8 expression was strongly correlated with immune cell infiltration across melanoma. The analysis revealed that higher expression of SLAMF8 was linked to greater immune cell presence in the tumor. Finally, the survival analysis was presented in Figure 5C, demonstrating that higher SLAMF8 expression and immune cell infiltration were associated with better overall survival in melanoma patients (p < 0.05).
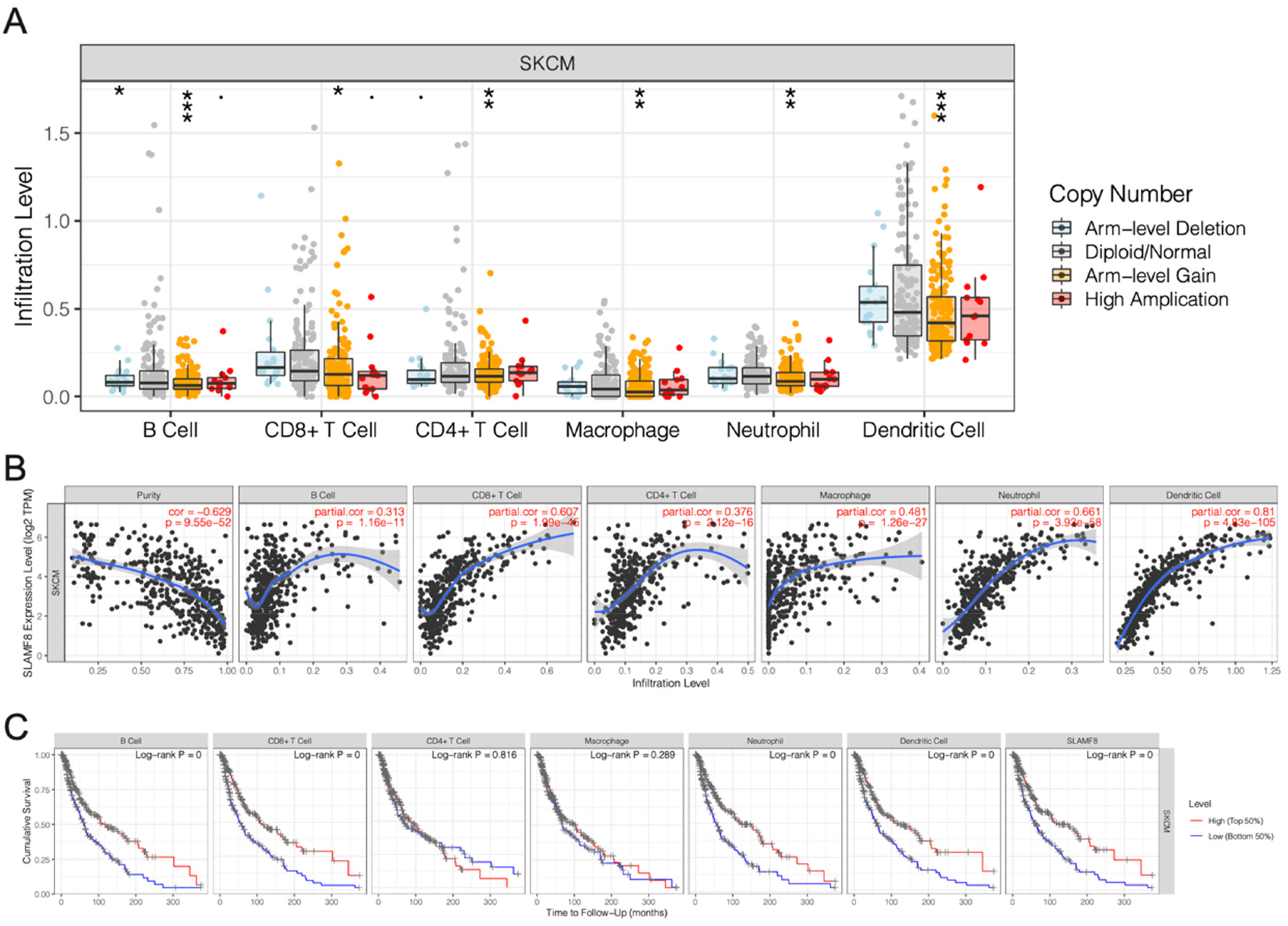
Tumor immune infiltration and its correlation with SLAMF8 in melanoma. (A) Analysis of somatic copy number alterations (SCNAs) in SLAMF8 reveals significant associations with immune cell infiltration levels in melanoma, suggesting a potential role in modulating the tumor immune microenvironment. (* p < 0.05; ** p < 0.01; *** p < 0.001). (B) SLAMF8 expression levels are positively correlated with the degree of immune cell infiltration, indicating that higher SLAMF8 expression is linked to increased immune presence within the tumor. (C) Kaplan–Meier survival analysis shows that patients with high SLAMF8 expression and elevated immune cell infiltration exhibit significantly improved overall survival (p < 0.05), supporting the prognostic relevance of SLAMF8 in melanoma.
SLAMF8-Related pathways and interactions in SKCM
The GSEA results revealed that SLAMF8 in SKCM is primarily associated with hallmark gene sets including allograft rejection, apoptosis, complement, IL2-STAT5 signaling, inflammatory response, TNFα signaling via NFκB, interferon gamma response, KRAS signaling, and IL6 JAK STAT3 signaling (Figure 6A). Subsequently, we constructed a protein-protein interaction network (Figure 6B), which included SLAMF1, LY9(SLAMF3), CD244(SLAMF4), SLAMF6, SLAMF7, SLAMF8, SLAMF9, FPR3, CD2, C1QB, and ARHGAP30, with most of these proteins belonging to the SLAMF family gene members. Furthermore, we performed KEGG and GO term enrichment analyses. The KEGG pathway analysis indicated significant enrichment in pathways such as Staphylococcus aureus infection, Pertussis, and Complement and coagulation cascades(Figure 6C). For GO biological process (BP) terms, significantly enriched categories included T cell activation (GO:0042110), natural killer cell activation (GO:0030101), positive regulation of type II interferon production (GO:0032729), T-helper 17 cell lineage commitment (GO:0072540), T-helper 17 cell differentiation (GO:0072539), regulation of type II interferon production (GO:0032649), T-helper cell lineage commitment (GO:0002295), positive regulation of cytokine production (GO:0001819), regulation of lymphocyte differentiation (GO:0045619), and positive regulation of interleukin-17 production (GO:0032740) (Figure 6D). In the cellular component (CC) category, enriched terms included collagen-containing extracellular matrix (GO:0062023) and intracellular membrane-bounded organelle (GO:0043231) (Figure 6E). For molecular function (MF), significant terms included complement receptor activity (GO:0004875), MHC class I protein binding (GO:0042288), MHC protein binding (GO:0042287), virus receptor activity (GO:0001618), SH2 domain binding (GO:0042169), and G protein-coupled peptide receptor activity (GO:0008528) (Figure 6F).
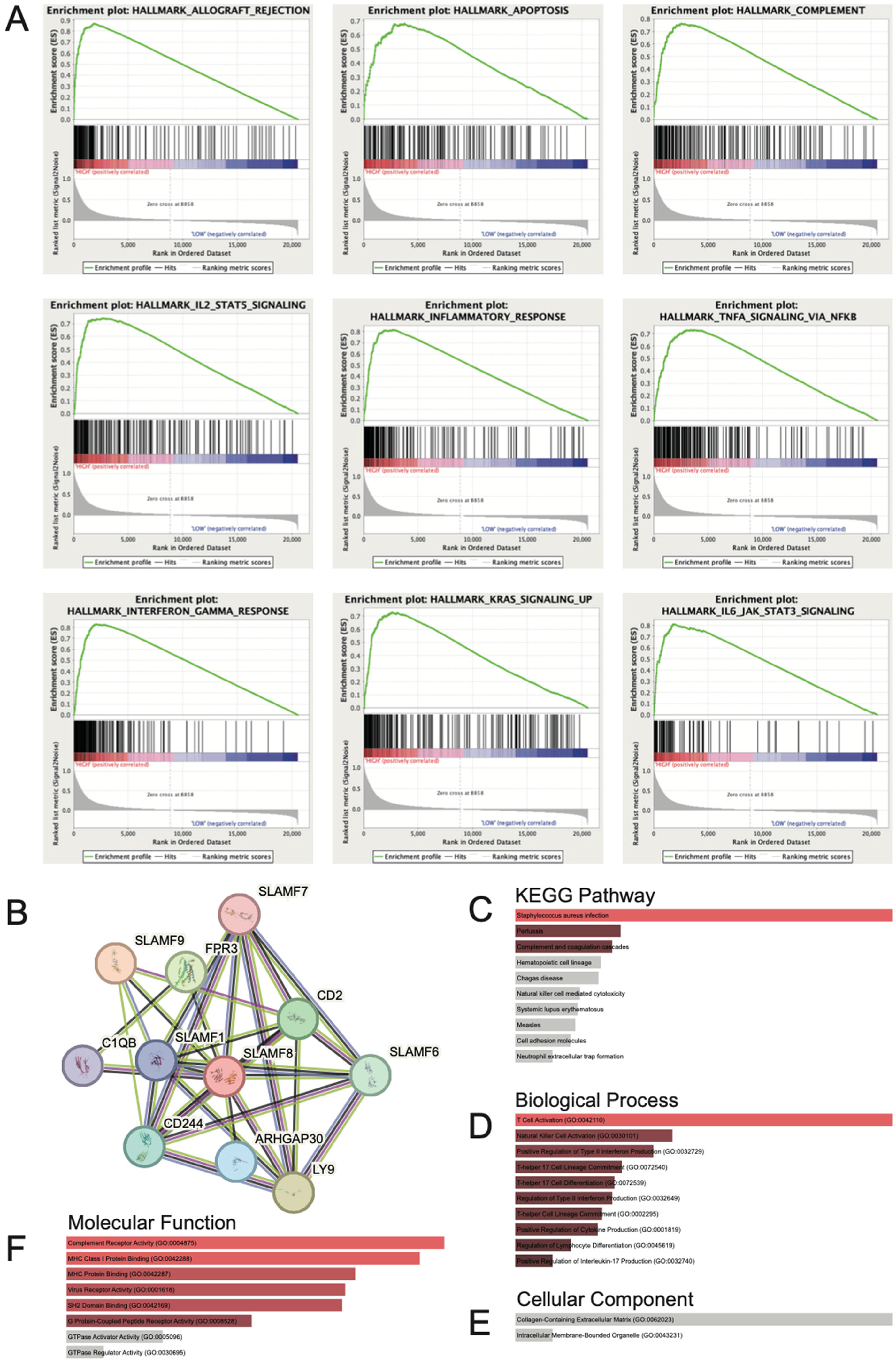
SLAMF8-Related pathways and interactions in SKCM. (A) Gene Set Enrichment Analysis (GSEA) indicates that high SLAMF8 expression is associated with hallmark pathways such as allograft rejection, apoptosis, complement, IL2–STAT5 signaling, inflammatory response, TNFα signaling via NFκB, interferon gamma response, KRAS signaling, and IL6–JAK–STAT3 signaling. (B) Protein–protein interaction (PPI) network constructed using STRING database shows SLAMF8 interaction with SLAMF1, LY9(SLAMF3), CD244(SLAMF4), SLAMF6, SLAMF7, SLAMF8, SLAMF9, FPR3, CD2, C1QB, and ARHGAP30, with most of these proteins belonging to the SLAMF family gene members. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis reveals significant involvement of SLAMF8-related genes in pathways including Staphylococcus aureus infection, Pertussis, and Complement and coagulation cascades. (D) Biological process is primarily enriched in T cell activation, NK cell activation, regulation of interferon and cytokine production, Th17 cell differentiation and lineage commitment, and lymphocyte differentiation. (E) Cellular component is mostly enriched in collagen-containing extracellular matrix and intracellular membrane-bounded organelles. (F) Molecular function is greatly enriched in complement receptor activity, MHC class I protein binding, virus receptor activity, SH2 domain binding, and GPCR peptide receptor activity.
Mendelian randomization analysis of SLAMF8 in SKCM
We employed a two-sample Mendelian Randomization approach to explore the potential causal link between SLAMF8 and melanoma. None of the selected SNPs exhibited characteristics of weak instrumental variables. The individual causal contributions of each genetic variant to SKCM are illustrated in Figures 7A and 7B. The Inverse Variance Weighted (IVW) analysis revealed a significant protective association of SLAMF8 with SKCM, yielding an odds ratio (OR) of 0.39 (95% CI = 0.21–0.74, p = 3.34e-03). Similarly, the Weighted Median approach corroborated this protective effect, with an OR of 0.40 (95% CI = 0.17–0.98, p = 4.52e-02). In contrast, the MR-Egger, Simple Mode, and Weighted Mode methods failed to demonstrate statistical significance (p > 0.05). A funnel plot analysis (Figure 7C) displayed a balanced distribution of effects, suggesting minimal bias, and the MR-Egger regression intercept indicated no evidence of horizontal pleiotropy (p = 0.26). A leave-one-out sensitivity analysis (Figure 7D) was conducted by iteratively excluding each SNP, and the consistent results across iterations confirmed that the observed causality was not driven by any single SNP, thereby validating the robustness of the MR outcomes.
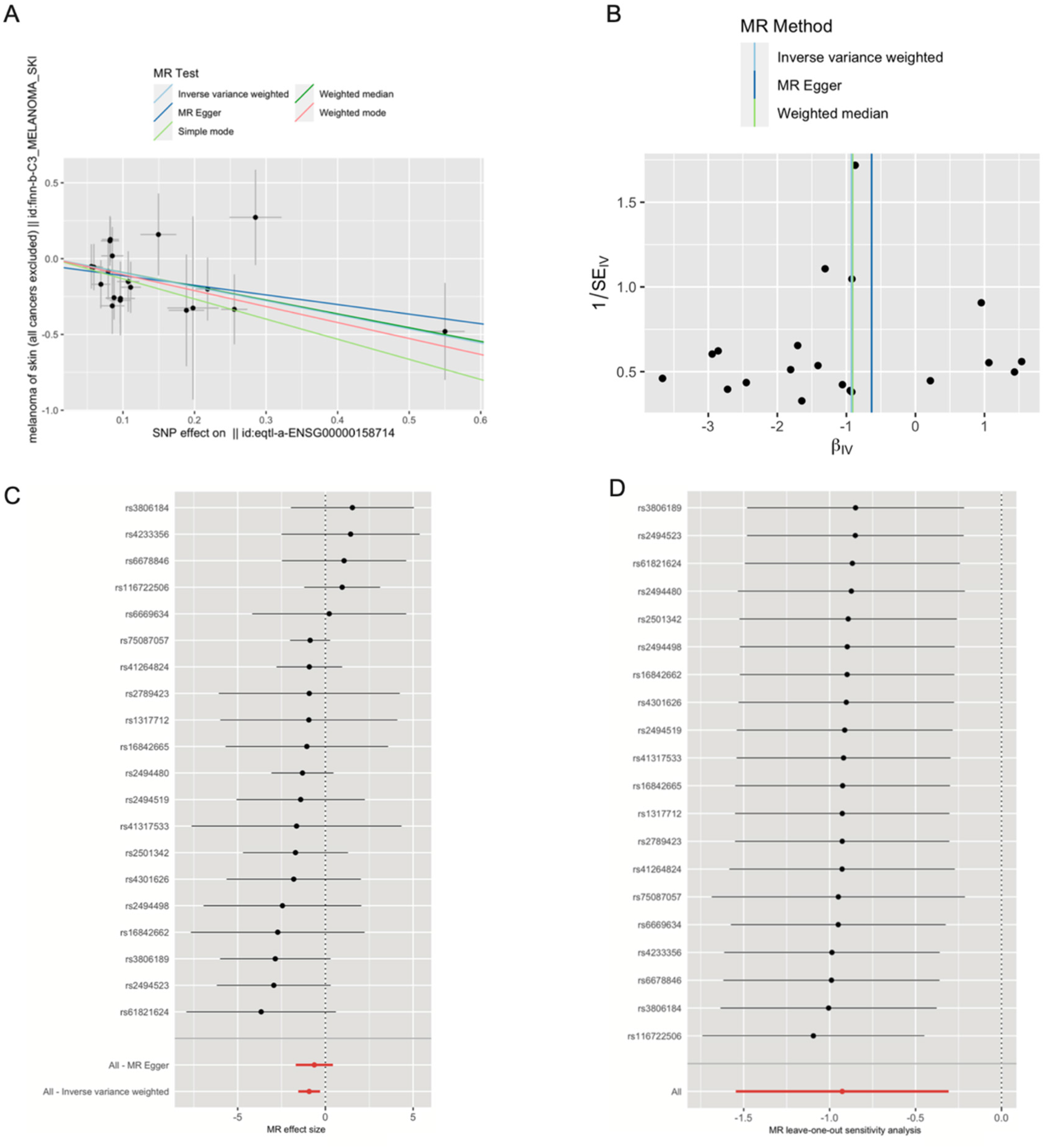
Mendelian randomization analysis of SLAMF8 and risk of SKCM. (A) Scatter plot showing the causal effect of SLAMF8 on the risk of melanoma. (B) Funnel plot visualizing the overall heterogeneity of MR estimates for the effect of SLAMF8 on melanoma risk. (C) Forest plot showing the causal effect of each SNP on the risk of melanoma. (D) Leave-one-out plot visualizing the causal effect of SLAMF8 on melanoma risk when leaving one SNP out.
Discussion
This study identified that SLAMF8 is significantly upregulated in malignant melanoma tissues, as well as in various other tumor types, including bladder cancer (BLCA), breast cancer (BRCA), cholangiocarcinoma (CHOL), esophageal carcinoma (ESCA), and glioblastoma multiforme (GBM) and so on. Notably, in melanoma, SLAMF8 expression was significantly higher in metastatic tumors compared to primary tumors and was also upregulated in tumor tissues compared to adjacent normal tissues. Further stratification by cancer stage revealed a nonlinear expression pattern, suggesting distinct roles for SLAMF8 at different stages of melanoma progression. Survival analysis demonstrated that higher SLAMF8 expression correlated with better prognosis in melanoma patients. Both DFS and OS analyses indicated that patients with high SLAMF8 expression exhibited significantly prolonged survival compared to those with low expression levels. To explore potential causality, we performed a two-sample MR analysis, which revealed a significant inverse association between genetically predicted SLAMF8 expression and melanoma risk. Although these findings suggest a protective role for SLAMF8, they should be interpreted with caution due to inherent limitations of MR analysis, such as horizontal pleiotropy and population stratification. Importantly, the functional role of SLAMF8 in melanoma was validated by in vitro experiments, which revealing that SLAMF8 suppresses the proliferation, migration, and invasion of melanoma cells. SLAMF8 knockdown significantly enhanced the migratory and invasive potential of melanoma cells, accompanied by an increase in PCNA expression. As PCNA is essential for DNA replication and cell cycle progression, its upregulation in SLAMF8-deficient melanoma cells suggests that SLAMF8 may negatively regulate proliferation-associated signaling pathways. One possible explanation is that SLAMF8 acts upstream of pathways such as MAPK/ERK or PI3 K/AKT, which are known to promote both PCNA expression and melanoma aggressiveness. Conversely, the reduction of PCNA observed in cells with higher SLAMF8 levels may reflect cell cycle arrest or a transition to a less proliferative state, implying that SLAMF8 might modulate melanoma cell behavior both directly and through its immunological role in the tumor microenvironment.
Furthermore, our study identified several key DNA methylation sites associated with SLAMF8 expression in melanoma. Specifically, five CpG sites exhibited a significant negative correlation with SLAMF8 gene expression, suggesting that DNA methylation may regulate its transcription. Survival analysis revealed that higher methylation levels at three of these CpG sites(cg07625783, cg06764092, and cg18084791) were significantly associated with worse overall survival, suggesting that epigenetic silencing of SLAMF8 may contribute to tumor progression and adverse clinical outcomes.
The mutational landscape of SLAMF8 in melanoma revealed genetic alterations in approximately 10.21% of patients, with missense mutations being the predominant type, which may impair SLAMF8 protein function or disrupt its immune-regulatory activities. Additionally, SLAMF8 SCNAs were strongly correlated with immune cell infiltration, indicating its role in shaping the tumor immune microenvironment. Higher SLAMF8 expression was linked to greater immune cell presence in the tumor, and patients with higher SLAMF8 expression and immune infiltration exhibited better overall survival.
GSEA analysis highlighted that SLAMF8 is primarily associated with immune-related pathways. The IL2-STAT5 signaling pathway is crucial for T-cell proliferation and activation, enhancing anti-tumor immunity. 27 Similarly, the IL6-JAK-STAT3 pathway plays a significant role in tumorigenesis and metastasis by promoting cancer cell survival, proliferation, and immune evasion. 28 The inflammatory response pathway contributes to the tumor microenvironment, influencing tumor growth and progression. 29 TNFα signaling via NFκB is involved in inflammatory processes and can affect apoptosis and cell proliferation in cancers. 30 The interferon-gamma response pathway has a dual role; it can enhance anti-tumor immunity but also contribute to immune resistance mechanisms. 31 KRAS signaling is implicated in cell proliferation and survival, with mutations often leading to uncontrolled cell growth in various cancers, including melanoma.32–35 Additionally, the PPI network analysis identified interactions with SLAMF family members and other immune-related proteins, supporting the hypothesis that SLAMF8 plays a key role in immune regulation.
Similar to PD-1, SLAMF8 is predominantly expressed on immune cells and can be upregulated by IFN-γ stimulation, shaping T cell–mediated immune responses. Across malignancies, its role appears context-dependent: in glioma, high SLAMF8 levels are linked to poor prognosis, chemotherapy resistance, and immunosuppression 15 ; in colorectal cancer (CRC), SLAMF8 correlates with lymphatic metastasis, immune checkpoint expression, and an immunosuppressive tumor milieu, while also predicting improved responses to anti-PD-1 immunotherapy in gastrointestinal cancers.16,36 These findings suggest that SLAMF8 modulates the tumor immune landscape, potentially by regulating macrophage plasticity, antigen presentation, and cytokine signaling.
SLAMF8 is predominantly expressed on tumor-associated macrophages (TAMs), which contribute to an immune-suppressive microenvironment. 17 However, our results indicate that melanoma cells themselves also express SLAMF8, implying that its presence is not solely driven by immune cell infiltration. Similar to SLAMF9, which is mainly found in TAMs but also in a subset of melanocytic cells, 37 SLAMF8 may have dual roles: as an immunomodulatory receptor on macrophages and as a signaling molecule intrinsic to melanoma cells. This dual origin resonates with observations in CRC, where SLAMF8 expression is strongly linked to immune infiltration and immunosuppressive signaling.16,36 In melanoma, a highly immunogenic tumor type where PD-1 blockade is a standard therapy, our findings suggest that SLAMF8 may serve as both a biomarker of immune activity and a cell-intrinsic modulator of tumor progression.
To our knowledge, this study is the first to comprehensively investigate the role of SLAMF8 in melanoma. Through experimental validation, multi-omics analysis, and Mendelian Randomization, we found that SLAMF8 expression is significantly upregulated in melanoma and associated with better prognosis. Its ability to suppress tumor progression, influence immune infiltration, and be regulated by DNA methylation positions it as a promising biomarker and potential protective role in melanoma. However, this study has several limitations that should be noted. The bioinformatics analyses were based on publicly available datasets, which may carry inherent biases such as sample heterogeneity and incomplete clinical information. While the Mendelian Randomization and in vitro experiments support a potential protective role of SLAMF8 in melanoma, further mechanistic studies and in vivo validation are needed to clarify its biological functions. Additionally, as a retrospective observational study, causal inferences should be interpreted with caution.
Conclusion
Our study demonstrates that SLAMF8 may serve as an important tumor suppressor in melanoma. Higher SLAMF8 expression correlates with improved patient survival, suggesting its clinical value as a prognostic marker. Mechanistically, SLAMF8 inhibits tumor cell proliferation and invasion, and its expression is regulated by DNA methylation. Mendelian randomization analysis further confirms its protective role against melanoma development. Additionally, SLAMF8 modulates immune cell infiltration in the tumor microenvironment. These findings reveal that SLAMF8 might serve as both a prognostic biomarker and a therapeutic target for melanoma treatment.
Supplemental Material
sj-docx-1-cbm-10.1177_18758592251392828 - Supplemental material for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study
Supplemental material, sj-docx-1-cbm-10.1177_18758592251392828 for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study by Jianjiang Liu, Wei Han and Guoliang Shen in Cancer Biomarkers
Supplemental Material
sj-docx-2-cbm-10.1177_18758592251392828 - Supplemental material for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study
Supplemental material, sj-docx-2-cbm-10.1177_18758592251392828 for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study by Jianjiang Liu, Wei Han and Guoliang Shen in Cancer Biomarkers
Supplemental Material
sj-docx-3-cbm-10.1177_18758592251392828 - Supplemental material for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study
Supplemental material, sj-docx-3-cbm-10.1177_18758592251392828 for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study by Jianjiang Liu, Wei Han and Guoliang Shen in Cancer Biomarkers
Supplemental Material
sj-xlsx-4-cbm-10.1177_18758592251392828 - Supplemental material for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study
Supplemental material, sj-xlsx-4-cbm-10.1177_18758592251392828 for SLAMF8 expression and prognostic significance in melanoma: A multi-omics and Mendelian randomization study by Jianjiang Liu, Wei Han and Guoliang Shen in Cancer Biomarkers
Footnotes
Author contribution
The work presented here was carried out in collaboration among all authors. Guoliang Shen defined the research theme, discussed analyses, interpretation, and revised the manuscript. Jianjiang Liu and Wei Han drafted the manuscript, analyzed the data, performed the experiments and interpreted the results. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets used and analyzed during the current study available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.