
Abstract
Non-small cell lung cancer (NSCLC) remains the most common cause for cancer-related mortality despite advances in treatment. Early detection is crucial for improving patient outcomes, yet current diagnostic and prognostic molecular biomarkers lack the sensitivity and specificity necessary to become clinically useful. Recent studies revealed that the lower airway microbiome play a role in NSCLC and that microbial signatures are associated with NSCLC development, progression, and prognosis, suggesting the potential for microbiome-based biomarkers for early diagnosis and risk stratification. Here we review recent advances in the role of the local and systemic microbiome in early-stage NSCLC. Primarily, several studies have identified specific microbial taxa associated with lung cancer suggesting novel insights into disease pathogenesis and progression. Integration of microbiome data with other ‘omics’ platforms, such as host transcriptomics and metabolomics, has the potential to enhance our understanding of microbial-host interactions and may provide more comprehensive biomarker signatures. While promising, challenges remain to the development of microbiome-based biomarkers such as those related to differences in samples utilized, sequencing methods, and data analysis. Here, we discuss such challenges as well as future directions for research needed to fulfil the promise of microbiome-based biomarkers for changing early detection and management strategies in NSCLC.
Introduction
Non-small cell lung cancer (NSCLC) remains the leading cause of cancer-related death in the world, representing 25% of all newly diagnosed cancers, and is estimated to cause 44% of total cancer-related deaths. 1 Early-stage NSCLC is curable with resection with 5-year survivals of 83%,65%, and 40% for TNM (Tumor, Node and Metastasis) stages I, II, and single station mediastinal nodal involvement, respectively. Nevertheless, only 29% of patients are found to have early-stage NSCLC. 2 Thus, there remains a critical need for new biomarkers that can aid the diagnosis of early-stage lung cancer. Importantly, NSCLC occurs in only a fraction of individuals eligible for lung cancer screening programs, and even if those screening program were successfully implemented, the adherence level is low which limits implementing widespread screening approaches for a large proportion of patients.3–5 Currently, blood-based biomarkers for the early detection of NSCLC lack sensitivity and specificity. Similarly, blood- or tissue-based biomarkers proposed to predict disease recurrence after potentially curative surgery lack sufficient accuracy to make them clinically relevant. Prognostication accuracy, which could determine whether a completely resected NSCLC could recur, is crucial for risk stratification to identify patients who may benefit the most from adjuvant therapy. Recently, with the growth of culture independent methods that allow for detecting microbial products in tissues and fluids there has been a new interest in exploring for microbial signatures in the setting of lung cancer, either by themselves or in combination with other ‘omics’, as a novel approach to develop biomarkers.6–11 In addition to the possibility of providing biological insights regarding lung carcinogenesis, this approach may help develop novel diagnostic and/or prognostic tools that ultimately could affect clinical decision-making. In the present review, we discuss recent advances in the study of microbiome of early lung cancer and its potential as diagnostic and prognostic biomarkers.
Airway microbiome and lung cancer pathogenesis
Traditionally, the lungs have been considered a sterile organ, but recent advances in sequencing techniques reveal that the lungs are commonly exposed to microbes that play a crucial role in immune regulation in normal lungs and may contribute to different disease processes.8,10 The majority of studies evaluating the microbiome in lung cancer have focused on characterizing differences between the microbiome of lung cancer and benign lung conditions, utilizing bronchoalveolar lavage (BAL) airway samples, and relying on 16S ribosomal RNA (16S rRNA) gene sequencing, a bacterial targeted approach to define taxonomic composition.10–13 Few studies have utilized a multi-omic approach combining microbiome, transcriptome, and possibly metabolome to uncover microbial signatures associated with lung cancer stage and prognosis.8,14–16
Several studies demonstrate that the lung microbiome is associated with carcinogenesis in NSCLC (Figure 1). Some of the studies have suggested that the microbiota of lung cancer is characterized by lower diversity compared to non-malignant conditions.11,17 Among the taxonomic associations described in lung cancer there is enrichment with several taxa commonly found as commensals of the oral cavity, such as Veillonella, Prevotella, Acidovorax, and Streptococcus.11,18 Other studies have described that lung cancer is associated with enrichment with pathogens, such as Staphylococcus. 10 Nejman et al. analyzed 1010 tumor samples and 526 paired surrounding normal tissue samples across seven tumor types (245 and 235 lung tumors and normal tissue samples) using 16S rRNA sequencing and found that in lung cancer there was a higher bacterial burden, measured by 16S rDNA copies, compared to negative control samples, and that intratumoral bacteria were mostly intracellular and present in both immune and tumor cells. Interestingly, that study showed that lung tumors were enriched with Proteobacteria, a major phylum of gram-negative bacteria that include many respiratory pathogens, such as Pseudomonas. 9 Tsay et al. reported that the lower airway microbiota of patients with NSCLC is enriched with several taxa commonly deemed to be oral commensals, such as Veillonella, Prevotella, and Streptococcus, when compared with disease controls. Those signatures were associated with dysregulation of pathways known to be involved in lung carcinogenesis, such as PI3K. 18 These findings were supported by experimental work demonstrating that exposure of KRAS-mutated epithelial cells to heat-killed bacterial products or supernatants of these taxa leads to upregulating Phosphoinositide 3-kinase (PI3K) and Extracellular signal-regulated kinase (ERK). In another study by Tsay et al., enrichment of the lower airway with oral commensal microbiota was associated with more advanced stage and disease progression, even among patients with early-stage lung cancer.17,18 Those observations were further expanded using a preclinical mouse model of NSCLC where introduction of some of the dysbiotic taxa, in particular Veillonella, led to a pro-inflammatory state with a T helper (Th)-17 signature and a more rapid disease progression.
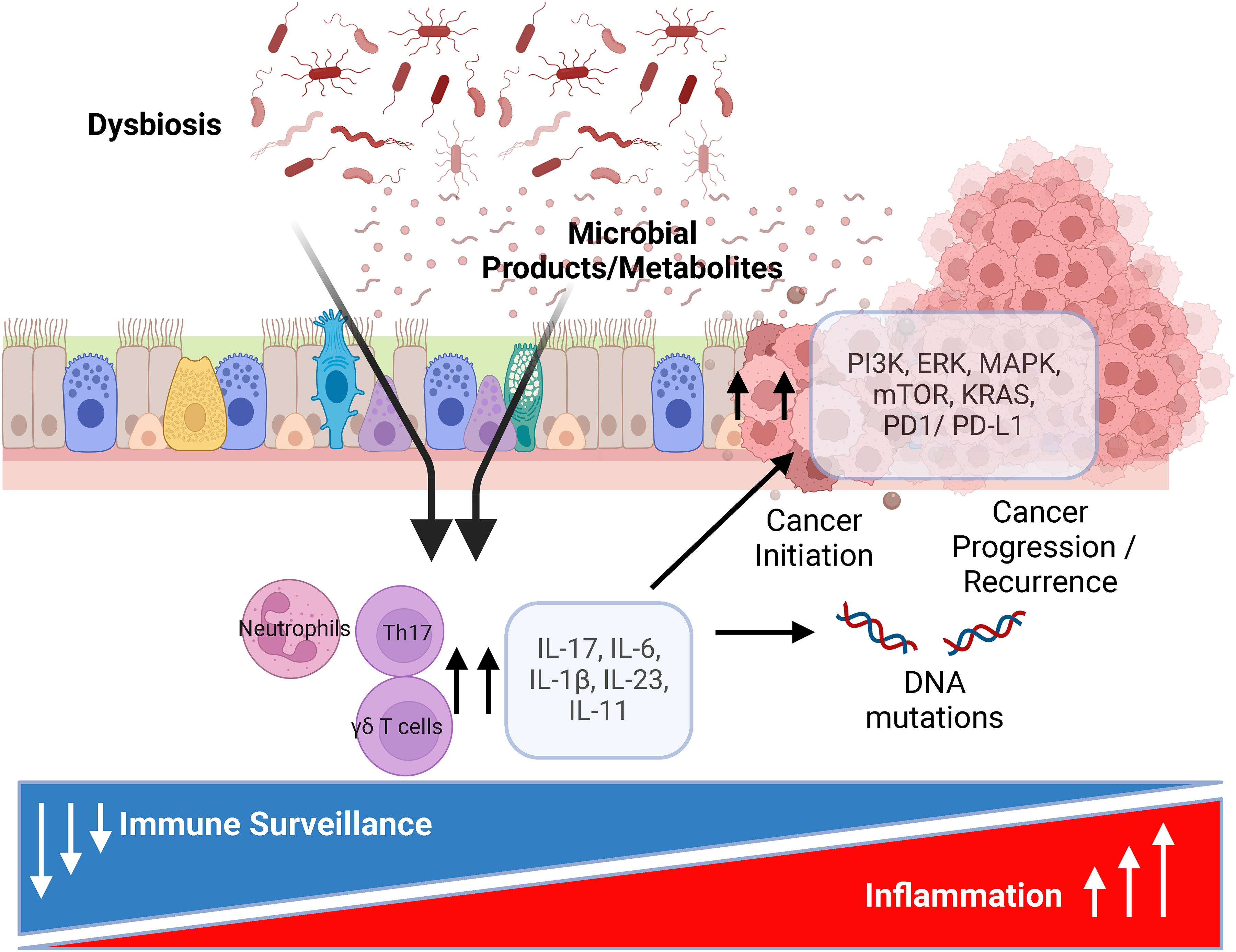
Lower airway microbiome and lung cancer pathogenesis. The microbiome plays a role in cancer development through the upregulation of pro-carcinogenic and inflammatory pathways. The microbiome modulates tumor microenvironment through different microbial products, such as metabolites, that may affect cancer progression through increased and persistent inflammation (Th17 mediated) and decreased immune surveillance that led to DNA damage and upregulation of pro-carcinogenic pathways.
In squamous cell carcinoma, tumors harboring TP53 mutation, a driver mutation for lung cancer development, have higher abundance of Acidovorax, another oral commensal. 19 Furthermore Stone et al., demonstrated in a preclinical mouse model that exposure to Acidovorax temperans accelerated tumor development and burden through infiltration of proinflammatory cells with interleukin (IL)-17T cell polarization. 20 The lung microbiome could potentially modulate other inflammatory pathways contributing to lung cancer progression. For instance, gram negative bacteria facilitate tumor progression through IL-33 pathway upregulation in NSCLC, 21 and the lung microbiome stimulate IL-1β production from myeloid cells. 22 IL-33 appears to have a dual role in lung cancer promoting tumor progression by inducing Th-2 cytokines on one hand, and enhancing CD8+ T cell activity, potentially inhibiting tumor growth on the other hand. 23
The findings described above align with data revealing that IL-17-producing γ-T cells, key modulators of the airway microbial-host interface, promote lung carcinogenesis after their enhanced activation by commensal lung microbiota, through the stimulation of Myd88-dependent IL-1β and IL-23 production from myeloid cells. 22 This Th-17 signature is associated with progression of lung cancer; interestingly, increased local and systemic IL-17, systemic IL-6, and higher neutrophil-to-T-cell ratio are associated with poor prognosis in lung cancer.24–27 PD-L1, the ligand for programmed death-1 (PD-1), is induced in non-lymphoid cells and tumor cells under inflammatory conditions triggered by several cytokines, such as interferon (IFN)-γ, and pathogen-associated molecular patterns (PAMPs).28–30 In addition, many signaling molecules (e.g., NF-κB, MAPK, PI3K, mTOR, and JAK/STAT) that affect proliferation, apoptosis, and cell survival induce PD-L1 expression.31,32 In a bi-transgenic mouse model expressing a conditional IL-17A allele and a conditional KrasG12D, increased IL-17 causes pro-tumorigenic inflammation that accelerates lung tumor growth and decreases survival. 33 Furthermore, Th-17 associated cytokines induce IL-11 release from multiple airway cells including eosinophils, bronchial fibroblasts, and epithelial cells.34–36 IL-11 is known to stimulate IL-33 expression in fibroblasts and may potentially have significant roles in cancer associated fibroblasts to influence tumor progression through modulating the tumor microenvironment in cancer. 37 Importantly, IL-11 can be upregulated by bacterial infection, promotes immune escape through PD-L1 upregulation in epidermal growth factor receptor (EGFR)-mutated NSCLC, and is a potential biomarker in NSCLC.38–40
Thus, the balance between Th17 inflammation and immune surveillance affects the response to immunotherapy in NSCLC. Further, the microbiome can influence cancer progression and response to therapy since an increase in pathogenic bacteria can lead to chronic inflammation through persistent generation of inflammatory mediators, thereby increasing the risk for DNA mutations and carcinogenesis.6,41 A growing body of evidence reveals that bacterial metabolites, such as short-chain fatty acids (SCFAs), are implicated in regulation of inflammatory tone in the lungs and could affect various signaling pathways and can cause direct DNA damage, thereby generating a pro-carcinogenic environment.41–44 All these findings reinforce that the local lower airway microbiome signature is lung cancer-specific, involved in lung cancer carcinogenesis, and contribute to lung cancer progression, and could be utilized as a biomarker in lung cancer diagnosis and progression.
Gut microbiome and lung cancer pathogenesis
Several investigations have now focused on how the gut microbiome can regulate the responsiveness to immunotherapy. For example, different gut microbiota signatures have been identified associated with augmented antitumor immunity and response to PD-1 blockade. Among lung cancer patients who did not respond to immunotherapy, the gut microbiota was characterized by low levels of Akkermansia muciniphila, and oral supplementation of this bacteria to antibiotics- treated mice restored the response to immunotherapy. 45 However, the mechanisms by which the gut microbiota affect lung cancer progression are not well established.45–47 While the gut microbiome role has not been extensively explored in lung cancer, some studies show that the gut microbiome can affect the lung through the gut-lung axis, and disrupt pulmonary immune homeostasis, and contribute to lung carcinogenesis through systemic inflammation and defective immunosurveillance. 48 However, whether other mechanisms such as microbial translocation in the gut is playing a role in the pathogenesis needs to be explored further.
The antitumor effect of Cytotoxic T-lymphocyte associated protein 4 (CTLA-4) monoclonal antibodies is altered by the gut microbiota, 49 and resistance to anti-CTLA-4 therapy may be due to loss of IFN-γ signaling. 50 IFN-γ is an important cytokine for host defense and its pathway is upregulated by many viruses and microbes. While gut microbiome signatures have been found to be associated with inflammatory tone and susceptibility to immunotherapy in melanoma and NSCLC, these data have been based on taxonomic identification and have been inconsistent among different studies.45–47,51 For example, Matson et al. found that in patients with melanoma, anti-PD-1 treatment responders had higher abundance of B. longum, C. aerofaciens, and E. faecium compared to non-responders. 47 In Gopalakrishnan et al., patients with higher bacterial diversity and increased relative abundance of Ruminococcaceae in the gut had enhanced systemic and anti-tumor immune responses. 46 Routy et al. identified that relative abundance of A. muciniphila was associated with clinical response to immunotherapy. 45 It is possible that individual taxa may not be as important as their functional aspects, such as their influence on specific metabolic pathways. These pathways which may be shared between different microbes, have significant immunological effects and could explain inconsistent taxonomic signatures among different cohorts. These investigations have led to clinical trials where the effects of fecal microbiota transplants are being evaluated to promote response to immunotherapy in resistant melanoma patients.52–54
The above shows that there is growing evidence showing that both airway and gut microbiome play a role in the pathogenesis of lung cancer. Whether microbial signatures can be identified and used to develop novel biomarkers for detection and prognosis of lung cancer is discussed below. But before such that discussion, we need to briefly introduce some methodological framework for microbiome data development (Figure 2).
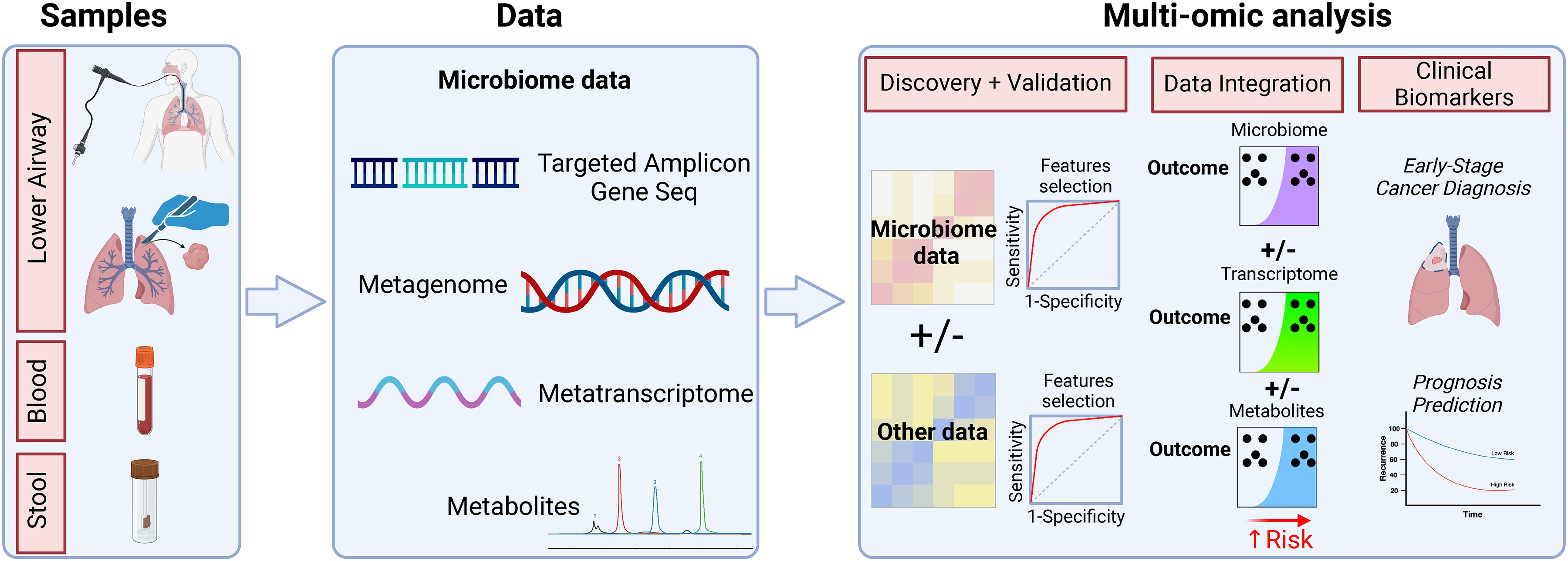
Potential use of multi-omics in developing microbial-based biomarkers for lung cancer. Multi-omics data may extend beyond taxonomic microbial data to microbial functions and host response data. Biomarkers developed and selected from microbial and host datasets should undergo validation. Multi-omics integration and analysis may uncover top features that can be used for lung cancer diagnosis and prognostication.
Special considerations about methodological challenges of developing microbial-based biomarkers
Sample selection for developing microbial-based biomarkers
Choosing the appropriate sample for biomarker development is an important consideration. Most airway microbiome studies in lung cancer rely on obtaining BAL, bronchial wash, sputum, or oral samples. 55 In addition, few studies have utilized tumor tissue or adjacent lung tissue to evaluate the microbiome.56,57 This is challenging given the importance of obtaining samples while avoiding DNA contamination of the harvested specimens at the time of surgery. In addition, for airway microbial biomarker development, while oral and sputum samples can be easily obtained, several studies show that these sample sites are not reflective of the microbial composition of the lower airways thereby limiting their potential as biomarkers.6,8,12,58 BAL with bronchoscopy has some advantages, including relative ease of sample procurement from the involved lobe or segment, and fewer complications than biopsies. However, performing bronchoscopies on large cohorts or longitudinally is challenging. 58 Exhaled breath condensate (EBC) might be seen as a promising non-invasive method for biomarker development, as it contains DNA and other molecules that can be used in biomarker discovery. However, two investigations comparing EBC samples to bronchoscopy samples suggest that EBC does not provide a reliable assessment of the microbial composition in the lower airways.59,60 For blood microbial biomarker development, blood samples allow for detection of cell free microbial DNA (cf-mbDNA). As with lower airway samples, these are extremely low biomass samples where detection of true microbial DNA is challenging, and might be affected by high levels of human DNA.by background DNA contamination, and by stochastic sequencing noise.60,61 While stool samples are accessible and easily obtained, the role of the gut microbiome for lung cancer biomarkers has not been extensively explored, and more studies are needed in this area.
Sequencing methods to obtain microbiome data
Most studies evaluating microbial differences between lung cancer and control samples profile the microbiome using sequencing of the 16S rRNA gene, a targeted approach seeking to amplify a short region of the bacterial genome containing variable regions that allow for taxonomic differentiation.9,11,13,17,56,57,62–65 16S rRNA sequencing is an easy, affordable, and now commonly used method to evaluate microbial signatures. However, there are some limitations to this method. 16S rRNA gene sequencing suffers from limited species resolution and focuses only on bacterial taxonomic assignment without allowing for functional assessment or evaluation of other non-bacterial fractions of the microbiome.10,58
Metagenomics and metatranscriptomics are high-throughput techniques that can characterize all different microbial fractions (based on broad base DNA or RNA sequencing, respectively) with potential for functional assessment of microbial genes present in the lung cancer microenvironment. 58 However, few studies thus far have used metagenomics to attempt to develop microbiome-based biomarkers in NSCLC.66,67 It is possible that these methods could provide further mechanistic insight regarding the role of the microbiome in early-stage lung cancer and aid in developing more robust and accurate biomarker signatures.
Recent advances in the microbiome research highlighted the importance of intratumor microbiota distribution within the tumor. Spatial transcriptomics showed that in colorectal and oral squamous cell carcinoma, the microbiome is highly organized in micro-niches between immune and epithelial cells, promoting cancer progression. 68 The integration of spatial transcriptomics with microbiome analysis offers an opportunity to identify distinct microbial signatures within tumor and normal adjacent lung tissues, which could lead to the identification of novel microbial biomarkers for early lung cancer detection.
Beyond the sequencing methods utilized, all these approaches are frequently challenging given the high risk for contamination and stochastic noise affecting the sequencing data.60,61 Several studies have documented these issues which are especially influential among low biomass samples such as those from the lower airways or blood.69–71 This risk of contamination by human or environmental genomic data might lead to significant doubts about prior findings. 72 Guidelines regarding how to develop and plan for studies involving low biomass samples such as the lower airways have been discussed elsewhere. 73
Another consideration when developing microbial signatures as biomarkers for NSCLC relates to the selection of cohort and outcome of interest utilized in the design of the study. Although this is not exclusive of microbiome biomarker development, we think that it is important to highlight here that each biomarker proposed is context dependent. For example, clear differences exist between developing a biomarker for diagnosis vs. a biomarker for prognosis. Similarly, diagnostic biomarkers developed comparing lung cancer vs. healthy controls may not necessary perform well when intended to use among a cohort of incidental lung nodules or among a cohort with significant smoking history with presence of a lung nodule. Finally, special confounders for microbiome studies such as antibiotic exposure and immuno-suppression are frequently present and difficult to fully control in investigations. These considerations will be discussed below as we present the current state of the art on microbial biomarker development.
Airway microbial biomarkers of lung cancer
The current understanding of the microbiome pathophysiology in lung cancer has promoted the investigation of the lower airway microbiome as a diagnostic and prognostic biomarker. Studies on lower airway microbiome have shown to help discriminate lung cancer patients from healthy controls. In a comparative study of 20 lung cancer patients with 8 patients with benign conditions, Lee et al. showed that Veillonella and Megaspheraere were relatively more abundant in lung cancer patients than control, and the combination of these two genera was able to predict lung cancer diagnosis with an area under the curve (AUC) of 88% (sensitivity = 95.0%, specificity = 75.0% and sensitivity = 70.0%, specificity = 100.0%, respectively). 11 Liu et al. compared 24 lung cancer patients with 18 healthy controls using protected specimen brushing samples, and reported that Streptococcus was significantly abundant among cancer patients and is able to discriminate lung cancer patients from healthy controls (AUC of 69.3%). 74 In another study including 25 lung cancer patients and 16 healthy controls, Bello et al. showed a high AUC of 89.7% using Streptococcus genera alone. 75 Jin et al. utilized metagenomics sequencing to study the lower airway microbiome among 91 lung cancer patients, 29 patients with non-malignant conditions and 30 healthy controls. 66 In their study, they show that 11 different types of bacteria, which were not previously identified, can help identify lung cancer from other conditions with an AUC of 79.6% (95% CI:67–92%). Cheng et al. evaluated the lower airway microbiome among 32 lung cancer patients and 22 patients with benign conditions, and developed a combined clinical tumor markers (CEA, NSE, CYFRA21-1) and bacterial markers (Pseudomonadaceae, Capnocytophaga, Stenotrophomonas, Microbacterium, Gemmiger, TM7-3, Oscillospira, Blautia, Lautropia, and Sediminibacterium) to distinguish lung cancer form benign conditions with an AUC of 84% (95% CI:74-94%). 76 Among the different taxa, the genera Pseudomonadaceae and Capnocytophaga contributed most to this classifier. In a similar study design, using machine learning, Kim et al. evaluated the lower airway microbiome in 24 lung cancer patients and 24 patients with benign conditions, and found that the lung microbiome can distinguish lung cancer form benign conditions with an AUC of 98%. 13 Most contributing taxa to the model were SAR202_clade and Acidobacterium.
The lower airway microbiome has also been examined for its utility for predicting cancer recurrence following surgical resection of early-stage lung cancer. Patnaik et al. compared microbial signatures in presurgical BAL samples in patients with stage I NSCLC, and observed that 19 genera were significantly abundant in 18 patients who had cancer recurrence within 32 months as compared to 18 patients without recurrence. Additionally, the presence of these microbial signatures predicted recurrence with an accuracy of 89% and an AUC of 77% (95% CI: 62–93%). 64 Peters et al. found that Clostridia, Bacteroidia (class), and Clostridiales, Bacteroidales (order) in non-involved adjacent tissue were associated with cancer recurrence in 46 patients with stage II NSCLC (43% of them had recurrence within 4.8 years).56,57 Using the relative abundance of Clostridiales and Bacteroidales, they constructed a microbial model predictive of recurrence which outperformed a model with standard covariates (age, sex, race, histology, smoking, and chemotherapy). Moreover, a combined model of these taxa with peripheral blood gene expression of IFITM2, TAP1, TAPBP, and CSF2RB, outperformed the models built using clinical, microbial, or gene expression data separately. These findings indicate that microbial signatures could be used to develop biomarkers that predict lung cancer recurrence among patients with early-stage resectable lung cancer.
While these studies have contributed new knowledge in this field, they are all limited by small size cohorts, the lack of external validation, and in some, absence of accounting for confounders such as histology, smoking status, treatment and the usage of medications (antibiotics, steroids). Furthermore, these studies were mostly performed in various lung cancer stage. For example, only 21.9% and 24.7% of cancer patients in Cheng et al. and Jin et al., respectively, were stage I lung cancer, while there were no patients with early-stage lung cancer in most of the other published investigations. These limitations can increase the heterogeneity of the results, which emphasize the need for standardized biomarker discovery and validation studies.
Gut microbial biomarkers of lung cancer
Interestingly, few studies have investigated the gut microbiome as a potential diagnostic biomarker for lung cancer. Zhang et al., reported that in stool samples, high abundance of gut microbes such as Bacteroides, Veillonella, and Fusobacterium were enriched among patients with lung cancer as compared to healthy controls (n = 41 for each group). 77 Zheng et al., using a discovery and validation cohort reported that the composition of the gut microbiome (beta diversity) differs between lung cancer patients and healthy controls. 78 Further, their analysis showed that a predictive model of 13 operational taxonomic unit (OTU) based biomarkers achieved high accuracy of lung cancer diagnosis (AUC = 97.6%). However, in their separate validation cohort of 34 lung cancer patients and 40 healthy controls the performance of this signature dropped significantly (AUC = 76.4%). These findings are promising, but larger investigations are needed to start understanding how the gut microbiome can be used as a biomarker for lung cancer.
Blood microbial biomarkers of lung cancer
The associations between circulating microbial signals and cancer diagnosis are not new. Indeed, observations made in the 1970s revealed associations between clinical bacteremia with Streptococcus bovis and subsequent diagnosed colorectal cancer (CRC). 79 This association was then validated and expanded to seven other bacterial species in a meta-analysis of more than 13,000 patients in Hong Kong, with concomitant hazard ratios up to 17.1 for later CRC diagnosis. 80 However, circulating microbial signals have not been associated with lung cancer until more recently when it was suggested that microbial signatures in peripheral blood using culture independent methods were found to be predictive of several cancer types, including lung cancer diagnosis. 67
In 2020, Poore et al. conducted one of the largest analyses of microbial DNA in cancer tissues and blood, enabling comparisons between dozens of cancer types. 67 These analyses included all treatment-naïve whole genome sequencing (WGS) and transcriptome data from The Cancer Genome Atlas (TCGA) seeking to identify the bacterial, viral, and archaeal nucleic acid content. The TCGA dataset comprised 18,116 samples from 10,481 patients across 33 cancer types. Of 6.4 × 1012 sequencing reads in TCGA, 7.2% were classified as non-human, of which 35.2% could be taxonomically assigned to bacteria, viruses, or archaea. The authors took several steps to limit the potential confounding effects of DNA signal contamination. Following different methods to identify potential DNA contaminants 92.3% of microbial sequence reads were discarded, and the remaining data were used for subsequent analysis. Using a machine learning approach, Poore et al. showed that tissue-driven microbial DNA or RNA features could effectively discriminate between one cancer type from all others (AUROCAvg = 97.3%, n = 32 tested cancer types). Moreover, microbial features from WGS in blood samples (cf-mbDNA) also revealed strong cancer type-specific discrimination (AUROCAvg = 97.2%, n = 20 tested cancer types), and similar diagnostic performances were exhibited when analysis were restricted to blood samples from patients with TNM stage I-II. These analyses revealed the presence of tissue-specific blood microbiomes in the context of cancer among 20 cancer types. Within the subset of samples from patients with lung cancer, the authors were able to identify a model that achieve an impressive predictive power to distinguish from healthy controls with an AUC of 97.2%. Poore et al. used as many as 1993 microbial genera, which is certainly a challenge for development of targeted approaches.
In a separate study by Nejman et al., microbes were profiled using an optimized 16S rRNA sequencing method on 1526 tumors and normal adjacent tissue from seven human cancer types with >800 experimental contamination controls. 9 They reached similar conclusions, identifying that microbes are ubiquitous among tumors and cancer-type specific. Together, the findings of Nejman and Poore indicate that a component of microbial DNA found in plasma may be tumor-driven and that these microbial signatures could be tissue-type specific. Another study utilizing digital droplet PCR (ddPCR) on 58 NSCLC patients and 58 healthy control individuals showed that the microbial abundance of three bacterial genera (Selenomonas, Streptococcus, and Veillonella) was higher in the blood of patients with NSCLC compared to healthy controls, and that these bacterial genera had a sensitivity of 75% and specificity of 78% for NSCLC diagnosis, regardless of stage or histology. 81 The performance of these biomarkers was confirmed in an independent validation cohort of 93 lung cancer cases and 93 controls. These studies are of interest in that several of these taxa were found to be enriched in the lower airways of patients with NSCLC by others. 17
Most recently, Chen et al., utilized WGS of plasma from 69 lung cancer patients and 97 healthy controls and found that the mean percentage of microbial reads was 0.012% in healthy controls and 0.009% in lung cancer patients. 82 Based on significant enriched species in the blood between lung cancer and healthy controls, they constructed a model for lung cancer diagnosis that achieved an AUC of 95% along with a sensitivity of 81%, a specificity of 90%, and an accuracy of 86%. The performance of the model was validated in two independent cohorts (overall 81 lung cancer and 68 healthy controls) revealing a combined sensitivity of 87% (95% CI: 78–93.6%), a specificity of 79% (95% CI: 67–87%), and an AUC of 93% (95% CI: 89–97%). Interestingly, one of the validation cohorts utilized was mostly patients with early-stage diseases, and there the model showed a high AUC of 92.1[86–97] %. In the same study, presurgical cf-mbDNA in the blood was evaluated as a biomarker for recurrence following surgical resection of lung cancer. To this end, 36 cancer patients who suffered recurrence within 3 years of surgery and 65 showed no recurrence and were included in the analysis. There, twenty-three taxa were identified as enriched in patients with recurrence, and 39 were enriched in the no recurrence group. Among the most enriched taxa in patients with recurrence investigators identified Candidatus family and Staphylococcus genus. A combined model using these microbial signatures exhibited a sensitivity of 71%, a specificity of 84%, and an AUC of 81%.
Overall, these studies are promising and support that microbial DNA signatures in peripheral blood may discriminate lung cancer from non-cancer conditions for early detection, with the potential for prognostic assessment after lung cancer treatment. However, the results of these studies should be interpreted carefully. As an example of the challenges faced with these data, the results of Poore et al. have recently been under scrutiny for the potential that human DNA contamination and technical artifacts may confound the results, 72 and led to the retraction of the initial study. This cautionary tale emphasizes the need for rigorous methodology and validation in these biomarker studies.
The microbiome and beyond for biomarker development
Microbes do not stand alone in the tumor microenvironment as they are constantly interacting with the host immune system and tumor cells.10,83 Therefore, a combination of different -omic approaches, such as host transcriptome and metabolomics, along with microbiome approaches could better evaluate the microbial-host interaction and help uncover the most promising biomarkers for early-lung cancer diagnosis and prognosis. Such integrative multi-omic approach for microbiome analysis might be critical for unveiling the repertoire of microbial features and their immunomodulatory functions that can play a significant role in lung cancer tumorigenesis and progression (Figure 2). Moreover, more studies are needed to evaluate whether this milieu of host-microbe interactions could lead to better diagnostic and/or prognostic performance than either one alone. 83 Additionally, multi-omic approaches may help better identify the most relevant biomarkers of diagnosis and prognosis, as such studies distill the most promising ones from among multiple microbial signatures, thus enhancing precision in biomarker development.8,16,17,18
An example of the integration of the microbiome with other omics includes the study of microbial metabolites. Zhang et al. examined microbiome and metabolites signatures in the lower airway of 28 lung cancer patients, while using ipsilateral non-tumor samples as controls, and revealed a collection of probiotic metabolite short-chain fatty acids (SCFAs) that was strongly associated with the tumor samples, and microbial species, such as Brachyspira Hydrosenteriae, were positively correlated with SCFAs. 84 While the study did not assess the performance of integrated data as a biomarker, it highlights the potential in multi-omic approaches. This integration approach for microbial signatures could be expanded also with radiomics features such as lesion morphology (solid, sub-solid, or ground glass), size, and FDG uptake level on PET scanning or other potential biomarker that may lead to a multi-pronged approach for biomarker development.
Challenges for the development of microbiome-based biomarkers and future directions
Although recent studies indicate that microbiome data has the potential to become diagnostic and prognostic biomarkers for lung cancer, some challenges still exist for its development. With growing interest in producing high quality data and influx of new technologies to improve microbiome biomarker development for early detection and prognosis prediction of lung cancer there are few challenges ahead. Several groups are currently looking at metagenomics and metatranscriptomics in NSCLC, as well as digital spatial approaches to evaluate the microbiome. Additionally, multi-omic approaches that incorporate metabolomics, genomics, and the microbiome are currently under development. More studies are needed to develop accurate microbial signatures, and these could then be integrated with evolving lung cancer radiomic imaging. Here are some other key important factors to consider for future research development include:
Cohort selection: It is crucial to select cohorts that are representative of populations where the biomarker is intended to be used. Thus, ensuring uniformity in lung cancer stage along with appropriate comparison groups, such as healthy controls, patients with non-malignant pulmonary nodules, and those with increased risk for lung cancer such as healthy smokers. Each of these might represent a different scenario for biomarker use and, since microbial composition is affected by many confounders, building well-balanced cohorts that account for confounders as sex, age, and smoking status is critical to minimize potential biases. For example, investigating the role of microbial biomarkers as part of lung cancer screening programs may be different than investigating those for the evaluation of indeterminate pulmonary nodules. Sample utilization and processing: While BAL samples are commonly proposed to conduct these investigations, bronchoscopy is an invasive procedure, a limitation when the biomarker is intended to be used at large population level, where noninvasive approaches are more desirable. Blood, EBC, and stool samples are more accessible and less invasive but more studies are needed to clinically validate them as biomarkers. The microbiome is affected by potential contaminants, particularly in low biomass samples such as lower airways and the blood. While recent studies on BAL are promising, more work is needed to ensure proper dealing with the challenges imposed by the low microbial biomass of these samples and the risk for background contamination and stochastic sequencing noise.60,61 Thus, these samples should be collected, stored, and processed within strict protocols to minimize variability and preserve microbial data integrity. DNA isolation should be optimized for low biomass samples. Current studies suffer from variability in sample selection and methods used to evaluate the microbiome. Therefore, future studies should adapt uniform sample utilized for biomarker studies, and standardized processing protocols. Analytical plan and validation of performance: Current studies suffer from lack of appropriate design for biomarker development. It is essential to build a discovery cohort then validating it on several validation cohorts, including internal and external validation. In addition, complex computational and bioinformatics pipelines are still needed to identify the most important features that can be used as biomarkers and integrate the data from different omic platforms. Most studies have evaluated the microbiome using 16 s technique. However, more advanced techniques such as metagenomics and metatranscriptomics are in principle more desired. Spatial transcriptomics are promising in their ability to characterize distinct microbial tissue niches that can be used to develop to identify potential microbial biomarkers. However, it is still not clear how these approaches will become the approach for biomarker testing or whether they are limited to the discovery phase where most promising candidates will be selected to then develop targeted approaches. Finally, a common challenge phased by these sequencing approaches represent the sparsicity and compositional nature of the data, which is well established for exploratory investigations.
85
However, quantitative approaches are the most commonly used for biomarkers and how to transition to such approaches when there are a large number of potential targets identified is still not very clear and needs further investigations.
In summary, there is growing literature supporting the role of the microbiome in lung cancer pathogenesis. This has spearheaded a series of investigations looking to harness on this new knowledge for biomarker development. Careful considerations addressing the challenges discussed here will be critical to fully assess the potential of the microbiome as a biomarker of lung cancer diagnosis and prognosis. Ultimately, there is a growing opportunity to leverage advanced technologies and multi-omics approaches to develop microbiome-based biomarkers that may lead to early detection, prognosis prediction and improved survival of patients with lung cancer.
Footnotes
Author contributions
CONCEPTION: FD, JCT, LNS, HIP.
INTERPRETATION OR ANALYSIS OF DATA: FD, JCT, LNS, HIP.
PREPARATION OF THE MANUSCRIPT: FD, JCT, LNS, HIP.
REVISION FOR IMPORTANT INTELLECTUAL CONTENT: FD, JCT, LNS, HIP.
SUPERVISION: LNS, HIP
Research support funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: U2C CA271890 (JCT, LNS, HIP, NIH/NCI), R37 CA244775 (LNS, NCI/NIH); R01 HL125816 (LNS, SBK, NHLBI/NIH); R56 HL151700 (MCK), PACT grant (LNS, FNIH); National Center for Advancing Translational Sciences (NCATS)
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.