
Abstract
Background
Among head and neck squamous cell carcinomas (HNSCCs), the incidence of oropharyngeal cancer (OPC) has been increasing in recent decades. Human papillomavirus (HPV) type 16 is associated with the majority of OPC. Circulating antibodies (Abs) to multiple HPV16 early antigens, including E2, E6, and E7, have been detected in patient sera, and are strongly associated with risk for OPC. However, HPV serology currently requires laboratory-based tests that are difficult to implement for large-scale cancer screening.
Objective
The goal of this study was to develop and validate a point-of-care assay for rapid detection of circulating IgG to HPV16 early antigens.
Methods
We measured Abs to HPV16 E2, E6, and E7 proteins using a lateral flow assay (LFA) in sera from 119 newly diagnosed OPC cases, 41 partners, and 81 healthy volunteers. The 119 patients with HPV-OPC were classified as HPV-positive based on in situ hybridization (ISH) or institutional p16 immunohistochemistry. The sensitivity and specificity of the LFA were determined by comparing to clinical diagnosis.
Results
The specificity for each individual HPV16 E2, E6, and E7 antibodies was 95.1% (77/81), 96.3% (78/81), and 98.7% (80/81), respectively. The sensitivities of the individual HPV16 antibodies were as follows: E2, 47.9% (57/119), E6, 31.9% (38/119), and E7, 57.1% (68/119). The 3-biomarker panel (at least one positive for E2, E6, and E7 Abs) demonstrated a sensitivity of 76.5% (91/119) and a specificity of 92.6% (75/81).
Conclusions
We developed a multiplexed lateral flow assay for the rapid detection of serologic responses to HPV16. Further research is required to determine the utility of these tests for HPV + HNSCC cancer screening, as higher specificity, and an assessment of the benefits of positive test results have yet to be evaluated in this context.
Introduction
Head and neck squamous cell carcinoma (HNSCC), including the oral cavity, oropharynx, larynx, and hypopharynx, is the seventh most common cancer worldwide. 1 In the United States, an estimated 54,000 new cases occurred in 2022. 2 About 70% of the patients with oropharyngeal squamous cell carcinoma are associated with human papillomavirus (HPV), and the incidence of HPV positive (HPV+) HNSCC is increasing. 3
Currently, there are no serology screening tests in clinical use for HNSCC. Physical examination is the primary approach for early detection of HNSCC, but the sensitivity (67%) and specificity (73%) are limited, and most patients present with nodal disease. 4 Oral HPV16 DNA is a potential biomarker, but the prevalence of up to 10% in the population and the rate of transient infections limit this approach.5–7
In contrast, serological screening has gained attention as a potential method for HNSCC screening in recent years. Antibodies to multiple HPV16 early antigens are detected in the majority of patients with positive HNSCC8–10 including prior to diagnosis.10,11 Seroconversion has been detected up to 28 years prior to diagnosis.10,12 Depending on the assay, the odds ratio (OR) of early antigen seropositive individuals developing HPVOPC is 400–500. 13 Another study showed that a significant proportion of HPV16-E6 seropositive individuals will develop oropharyngeal cancer (OPC), with 10-year risks ranging from 17% to 27% for males and 4% to 6% for females aged 50–60 years in the United States 14 Given the substantial predictive power of HPV16 serology, there is a clear need for rapid, cost-effective methods to detect HPV16 antibodies for population-based screening HPV positive HNSCC and for identifying high-risk individuals for monitoring studies.
HPV16 E2, E6, and E7 are early proteins involved in the viral life cycle and contribute to oncogenesis. E6 and E7 are oncogenes that play a direct role in cancer progression by disrupting tumor suppressor pathways, while E2 regulates the viral genome and can influence the expression of E6 and E7.15,16 These proteins are key targets for the immune system and are frequently recognized in serologic assays for HPV-associated cancers.11,17
There are several challenges to point-of-care serology for HPV16. First, the protein stability of the HPV16 E6 protein has limited its successful integration into assays. 18 Second, based on our experience, the heterogeneity of the serologic response to HPV suggests that multiple biomarkers will be necessary for effective screening. 8 Third, the low titers of antibodies limit the utility of traditional colorimetric LFAs.
To address these challenges, we systematically tested the core immunogenic fragments of E6, E2, and E7 to develop one of the first multiplexed LFA for HPV serology. 19 Using well-validated serum samples from the HOTSPOT study of participants with newly diagnosed HPV-related OPC (HPVOPC), their partners, and healthy controls,8,20 we developed a 3-Ab panel for HPVOPC detection.
Materials and methods
Serum and whole blood sample collections
The sera from 119 cases, 41 partners, and 81 healthy volunteers were derived from the multicenter HOTSPOT (Human Oral Papillomavirus Transmission in Partners over Time) study which has been previously reported.8,20 Written informed consent was obtained from all subjects under institutional review board approval. Clinical Trials number: NCT01342978. Fingerstick blood samples were collected from healthy volunteers after written informed consent and approval by the Institutional Review Board (IRB) at ASU (STUDY00002869).
Fluorescent microsphere conjugation
Carboxyl-modified fluorescent F1Y050 microspheres (#FR180380534, EMD Millipore) were conjugated to Goat anti-human IgG (#109-005-008, Jackson ImmunoResearch Laboratories) using a two-step EDC (#22980, Thermo Scientific) / Sulfo-NHS (#24510, Thermo Scientific) covalent coupling procedure. The details of this process have been previously described by Millipore. 21
Expression and purification of HPV16 proteins
The aim of this process was to produce and purify recombinant fragments of HPV16 E2, E6, and E7 proteins to be used for subsequent serological analysis and development of diagnostic assays. The N-terminus of HPV16 E6 (
Serologic recognition of recombinant proteins by ELISA
To evaluate the antigenicity of the recombinant proteins, 96-well plates were coated overnight at 4 °C with the recombinant proteins (20 ng/well for HPV16 E7 and HPV16 CE2, 40 ng/well for HPV16 E6). Plates were subsequently washed 5 times with PBS (#BP6651, Fisher Scientific) 0.2% Tween 20 (#P1379-1L, Sigma-Aldrich) (PBST) and blocked with 200 µl of 5% (wt/vol) skim milk (0290288725, MP Biomedicals) in PBST (0.2%) for 1 h at room temperature. The wells were washed 5 times with PBST (0.2%) and incubated with 100 µl serum dilutions pre blocked with Escherichia coli lysate and 0.2% milk-PBST overnight. 22 After 1 h of incubation with sera, plates were then washed 5 times with PBST (0.2%) and incubated with anti-human IgG antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:10,000 for 1 h. The wells were washed 5 times with PBST (0.2%) after 1 h and detected the signals using Supersignal ELISA Femto chemiluminescent substrate (#37074, Thermo Scientific). Luminescence was detected as relative light units (RLU) on a Glomax 96 microplate luminometer (Promega, Madison, WI) at 425 nm. The ratio of RLU for individual HPV-specific antibodies to RLU for the control GST-protein was measured.
Lateral flow assembly and assay using finger-stick samples
To assess the configuration of the lateral flow assay for HPV serology testing, we first used EBNA-1 as a model antigen. 114 μg/mL of EBNA-1 (#MBS318777, MyBioSource), 114 μg/mL of BSA (#30-AB74, Fitzgerald), and 300 μg/mL of human IgG (#102643-682, VWR) proteins were dispensed by an automated lateral flow reagent dispenser (Claremont Biosolutions) on three sites spaced at 6 mm intervals on the same nitrocellulose (UniSart CN95). A 7 mm Vivid GX plasma separation membrane (#NC1557225, Fisher Scientific) was attached to a 9 mm sample pad (#CFSP223000, EMD Millipore Corp) followed a 40 mm nitrocellulose and a 27 mm absorbent pad (#8117-2250, GE Healthcare). To perform the assay, 60 μL of PBST (0.2%) was added to pre-wet the LFA strip. 5 µl of healthy donor whole blood was added to the plasma separation membrane, chased with 60 µl PBST (0.2%) for 25 min, and strips were washed in two steps with 50 μL of PBST each. Then 60 μL of diluted anti-human IgG conjugated with F1Y050 microspheres (5.74 × 107 estimated number) were suspended in BlockAid (#B10710, Invitrogen)/PBST, sonicated in a bench top sonicator (#CPX1800, Ultrasonic Cleaners) for 2 min, and subsequently applied to the strips (30 μL × 2). After 20 min, the strips were washed by applying 100 μL of PBST (0.2%) to the strip (50 μL × 2). The strips were then dried on a hot plate at 37 °C for 10 min. The results were assessed by detection in a custom point-of-care fluorescent reader (see below, and 23 ).
HPV16 LFA
The aim of this section was to optimize the LFA for detecting HPV16-specific antibodies and assessing assay sensitivity by serial dilution of anti-HPV16 E7 monoclonal antibody.
25 μg/mL of BSA, 1 mg/mL of HPV16 NE6, 0.5 mg/mL of HPV16 CE2, 0.5 mg/mL of HPV16 E7, and 0.4 mg/mL of human IgG proteins were dispensed on the test lines and control line using a dispenser (XYZ3060TM, Biodot) (Figure 1(a)). Nitrocellulose membrane sheets and the absorbent pads were assembled then cut into 50 mm × 5 mm strips by a specialized cutter (Matrix 2360, Kinematic Automation) and mounted on glass slides (#16004-422, VWR) with adhesive (#GBL620001-1EA, Sigma-Aldrich). The slides were dried in a desiccator overnight before use.
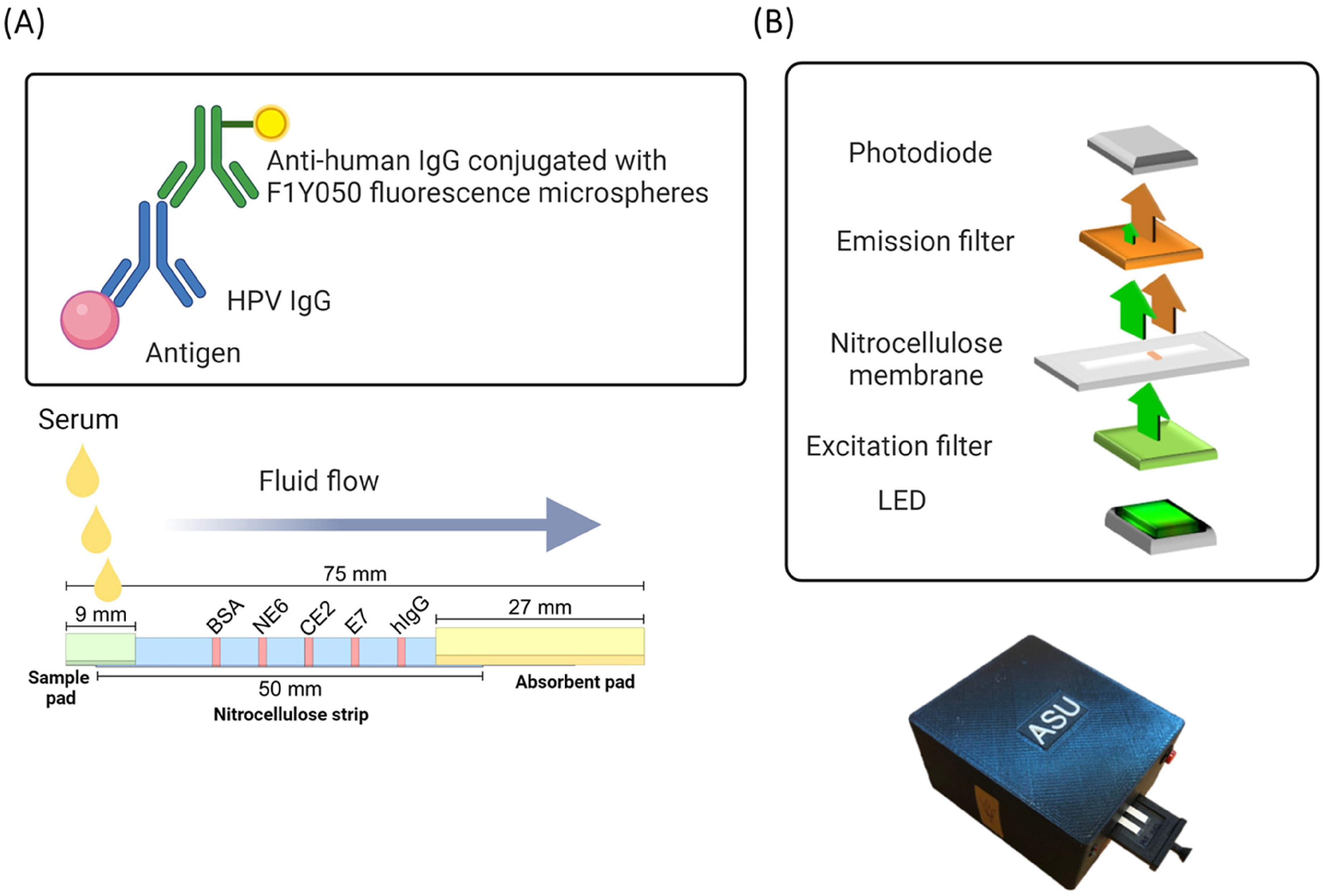
Design of lateral flow assay. (A) BSA, HPV16 antigens NE6, CE2, E7, and human IgG were immobilized at 6 mm intervals on a nitrocellulose strip. After flow of plasma across the strip, IgG was detected with anti- human IgG conjugated F1Y050 fluorescent microspheres. (B) Circuit schematic inside the reader showing charge-integration amplifier readout circuit, biorecognition sites on a microscope slide and LEDs used as the excitation source. 23
For the limits of detection, a serial dilution of anti-HPV16 E7 monoclonal antibody (#MCA2818, Bio-Rad) ranging from 0.01 ng/mL to 10 μg/mL were spiked into negative control serum. To prepare the LFA, 50 μL of PBST (0.2%) was added to pre-wet the strip. Subsequently, 30 μL of diluted monoclonal antibody was added upstream of the strip, allowing it to flow through the strip by capillary action for 15 min. After 15 min, the strips were washed by flowing 100 μL of PBST through the test strip in two steps of 50 μL each aliquot. Then 60 μL of diluted anti-human IgG conjugated with F1Y050 microspheres (5.74 × 107 estimated number of microspheres) suspended in BlockAid /PBST and sonicated in a bench top sonicator for 2 min, then dispensed onto the strips in two steps of 30 μL each aliquot and allowed to flow through for 20 min. The strips were washed with 100 μL of PBST in two steps of 50 μL each aliquot. After the washing steps, the strips were dried on a hot plate at 37 °C for 10 min. The results were assessed by detection in a point-of-care fluorescent reader. 23 Serum samples were diluted 1:50 in PBS, following the same protocol as described above.
Point-of-care fluorescent reader
This section describes the design, assembly, and function of the point-of-care fluorescent reader used to detect fluorescent signals from the LFA. The details of the reader design, construction, and operation have been described in detail in previous paper. 23 Briefly, the reader uses a combination of LEDs (light emitting diodes) and optical filters to create an inexpensive handheld fluorimeter. The fluorescence is detected with a photodiode that integrates the photocurrent to produce a voltage correlated to the fluorescence intensity. This reader is a universal fluorescent sensor and compatible with different wavelengths, depending on the excitation and emission filters used. 23 It was specifically designed for applications in low and middle-income countries (LMICs) with clinical utility in mind. The device operates on battery power with minimal fluidics and requires minimal user training and currently connects to a laptop. The readout data is entered into an Excel file, which automatically calculates the Signal-to-Noise Ratio (SNR) based on the target site and negative site output within the same strip.
Quality control of LFA
To evaluate the sensitivity of LFA, case serum samples and a control sample were prepared at concentrations of 1:10, 1:50, 1:250, 1:1250, and 1:6250 and tested. The fluorescence signal intensities for HPV16 NE6, HPV16 CE2, and HPV16 E7 were measured using the fluorescent reader. The SNR of IgG was determined by dividing the signal intensity of each specific HPV16 protein by the signal from the BSA control protein in serum samples. Each serum sample was tested in triplicate to ensure accuracy and reproducibility.
Next, to determine the lower limit of detection, BSA (25 μg/mL), HPV16 E7 protein (0.5 mg/mL), and human IgG (0.4 mg/mL) were dispensed onto the negative control, test, and positive control lines, respectively, on LFA strips. Serial dilutions of a monoclonal HPV16 E7 antibody were spiked into negative plasma and tested to determine detection limits. Each dilution was tested in triplicate to ensure repeatability and precision. Signal intensity for each test was measured using the fluorescent reader. The limit of detection (LOD) was determined by comparing the signal intensity of the negative control line (BSA) with that of the test line (HPV16 E7).
To evaluate long-term stability of LFA, the strips were sealed with desiccants under vacuum and stored at room temperature (25 °C). The assays were performed monthly over the course of four months to monitor protein stability.
Statistical analysis
We compared the SNR for each of the HPV 16 antibodies between the case, partner, and healthy control groups using the Mann-Whitney test. We defined a positive antibody response as greater than the mean SNR plus two standard deviations from the control samples and compared HPV 16 antibody prevalences between case and control groups using Fisher's exact test. We then performed the receiver operating characteristics (ROC) curve analysis for the HPV 16 antibodies with area under the ROC curve (AUC), odds ratio, sensitivity at 90% specificity and Fisher's exact test. We constructed a panel of the HPV 16 antibodies using LASSO regression and evaluated the improvement in diagnostic performance using fitted and leave-one-out cross-validation ROC curves. P-values less than 0.05 were considered statistically significant.
Statistical analyses were performed using R version 4.0.0 and GraphPad Prism version 10.
Results
Design of a multiplexed lateral flow fluorescent HPV16 serologic assay
The focus of this study was to develop a LFA for the detection of HPV16 early-antigen-specific IgG in the blood of participants with HPVOPC. From on our previous laboratory-based ELISA studies using mammalian in vitro translated proteins, we predicted that a panel of three antigens (E2, E6 and E7) would be sufficient to achieve >70% sensitivity of detection of cases at diagnosis at >90% specificity. 24 We made multiple modifications to existing lateral flow systems to accommodate the multiple antigens, challenges of protein expression and stability, and the low titers of IgG detectable in sera. First, we designed a system to print multiple antigenic lines on a single lateral flow membrane (Figure 1(a)). Second, we used anti-IgG fluorescent microspheres for detection, which increased the lower limits of detection but also increased the number of required washing steps (Figure 1(a)). Third, we developed a point-of-care fluorescent reader using LEDs with a charge-integration amplifier circuit, designed for resource-limited settings (Figure 1(b)). 23 To use the system with fingerstick blood collection, we chose an inexpensive, rapid, easy-to-use and disposable filter based microfluidic plasma separator (Vivid GX plasma separation membrane) which uses an asymmetric vertical membrane to filter plasma from whole blood samples. This captures the cellular components of the blood (red cells, white cells, and platelets) in the large pores and allows plasma to flow through the smaller pores on the downstream side of the membrane.
To evaluate the printing, detection methods and the platform, we first measured anti-EBNA-1 IgG. The EBNA-1 protein is highly immunogenic in >90% of healthy US blood donors. We printed purified viral EBNA-1 protein, negative control (BSA), and positive control (human IgG) proteins. We optimized the plasma separation membrane, protein printing, incubation times and wash steps (data not shown). We measured anti-EBNA-1 IgG from a finger-stick of blood (5 μl) for 20 healthy subjects. Intra- and inter-assay variability were measured between slides and printing batches and ranged from 1.2–25.1% (Supplementary Table 3).
Generation of recombinant, immunogenic HPV proteins
LFAs require large quantities of purified proteins, ideally suited for bacterial expression. The purification of HPV16 E6 protein from bacteria is challenging due to the three predicted hydrophobic regions of HPV16 E6, amino acids 31–36 (IHDIIL), amino acids 58–62 (LCIVY), and amino acids 104–108 (LCDLLI). 25 One approach is to co-transfect an HPV16 E6 expression vector and the chaperone plasmid into E. coli to generate soluble protein. 18 Another approach is to delete one hydrophobic amino acid region 31–36 (IHDIIL) with expression in the pGEX4T1 vector. 25 We mapped immunogenic fragments of E6 expressed in the pDEST15 vector without co-transfection of chaperone plasmid. The first two constructs express a deletion hydrophobic region of HPV16 E6 (Δ31–36 aa), 25 and the region of amino acids 70–100 which excludes these three predication hydrophobic regions. A different region (91YGTTL95) of HPV16 E6 is a highly conserved epitope (YGD/XTL) among HPVs E6 which has been identified as a B-cell epitope in HPV58 E6. 26 Therefore, we truncated HPV16 E6 into two regions. One is HPV16 CE6 which contains the highly conserved epitope (91YGTTL95), and the other one was HPV16 NE6. The IPTG induced expression of different versions of HPV16 E6 proteins and other HPV16 CE2 and HPV16 E7 were in fusion with the N-terminal GST and confirmed by SDS–PAGE (Figure S1).
First, we investigated the background reactivity to these different versions of HPV16 E6 proteins using control serum in a laboratory ELISA format. HPV16 E6 (Δ31–36 aa), NE6, E6 (70–100 aa), and CE6 GST fusion proteins were coated on a 96-well plate and detected using serial dilutions of control serum sample and anti-human IgG antibody conjugated with HRP. From Figure 2(a), CE6, E6 (70–100 aa), and E6 (Δ31–36 aa) have strong background signals for the control serum. HPV16 NE6 was selected and then tested using a serial of dilutions of control and case serum samples by ELISA (Figure 2(d)).
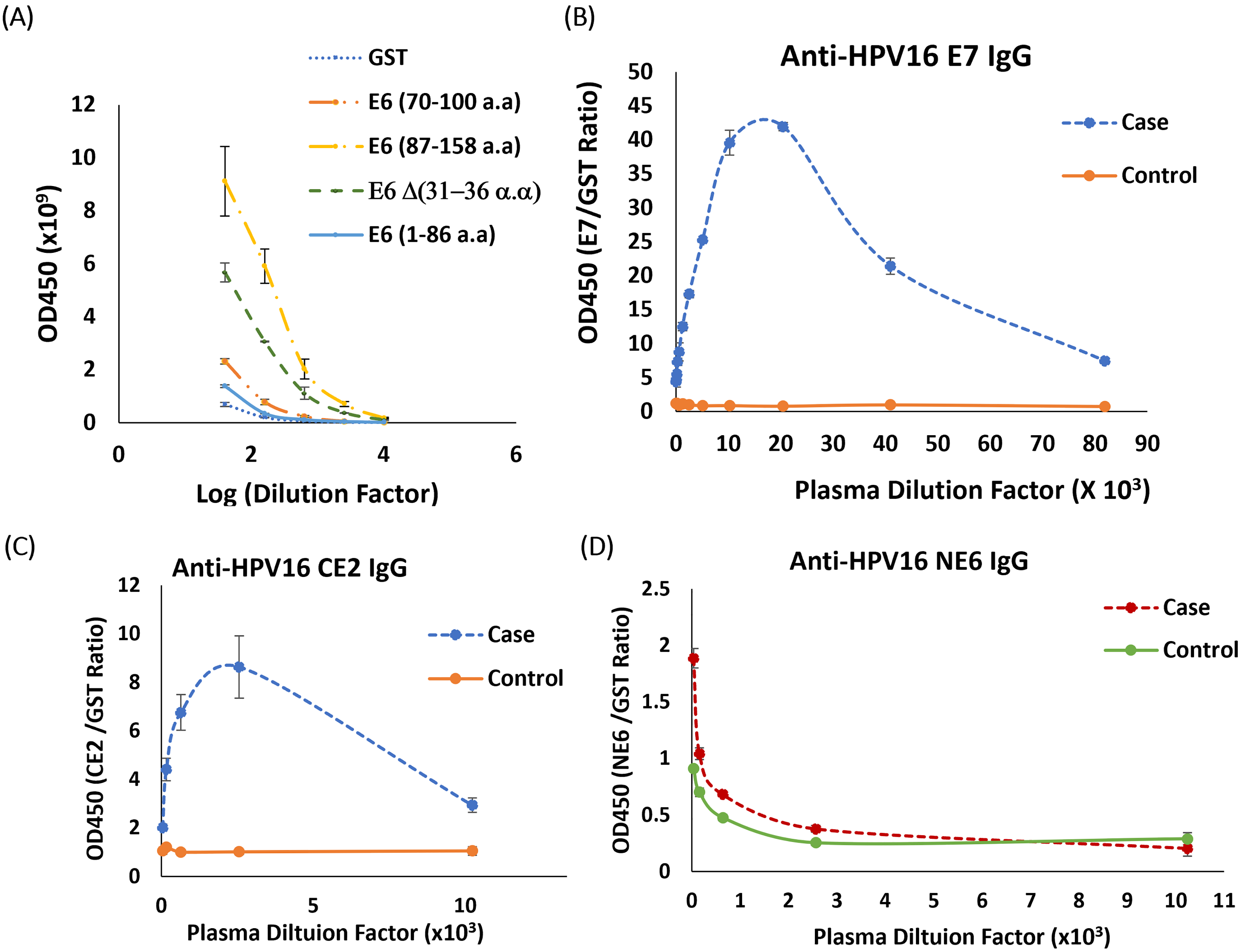
Comparison of antigenicity of HPV16 recombinant proteins and polypeptides. (A) Different fragments of HPV16 E6 (B) HPV16 E7 (C) HPV16 CE2 (D) HPV16 NE6 recombinant proteins were coated on a 96-well plate (20 ng/well for HPV16 E7 and HPV 16 CE2, 40 ng/well for different fragments of HPV 16 E6 and incubated with serially diluted case and control sera, followed by anti-human IgG HRP antibody at 1:10,000 and detected using Supersignal ELISA Femto chemiluminescent substrate. Luminescence was detected as RLU at 425 nm. The ratio of RLU for individual HPV-specific antibodies to RLU for the control GST-antigen was measured. Each HPV16 protein ELISA analyses were performed in triplicate. Error bars represent 95% confidence intervals.
Next, we purified recombinant HPV16 CE2 and full-length HPV16 E7 as N-terminal GST fusion proteins and tested the protein products by ELISA. From Figure 2(b) and 2(c), both HPV16 CE2 and HPV16 E7 proteins were selectively detected using case sera. The final 3-antigen panel (HPV16 NE6, HPV16 CE2, and HPV 16E7) were chosen for the multiplexed LFA.
Quality control of the LFA
The proteins BSA (negative control), HPV16 NE6, HPV16 CE2, and HPV16 E7, and human IgG (positive control) were dispensed on five sites spaced at 6 mm intervals on the same nitrocellulose strip (Figure 1(a)) and assembled for testing. As shown in Figure S2, all three antibodies could be detected within the ranges of 1/50 to 1/250 dilution. To measure the lower limits of detection, we conducted a serial dilution of anti-HPV16 E7 monoclonal antibody ranging from 0.01 ng/mL to 10 µg/mL, which was then spiked into negative control serum. We detected at least 10 ng/mL of anti-HPV16 E7 antibodies (Figure 3).
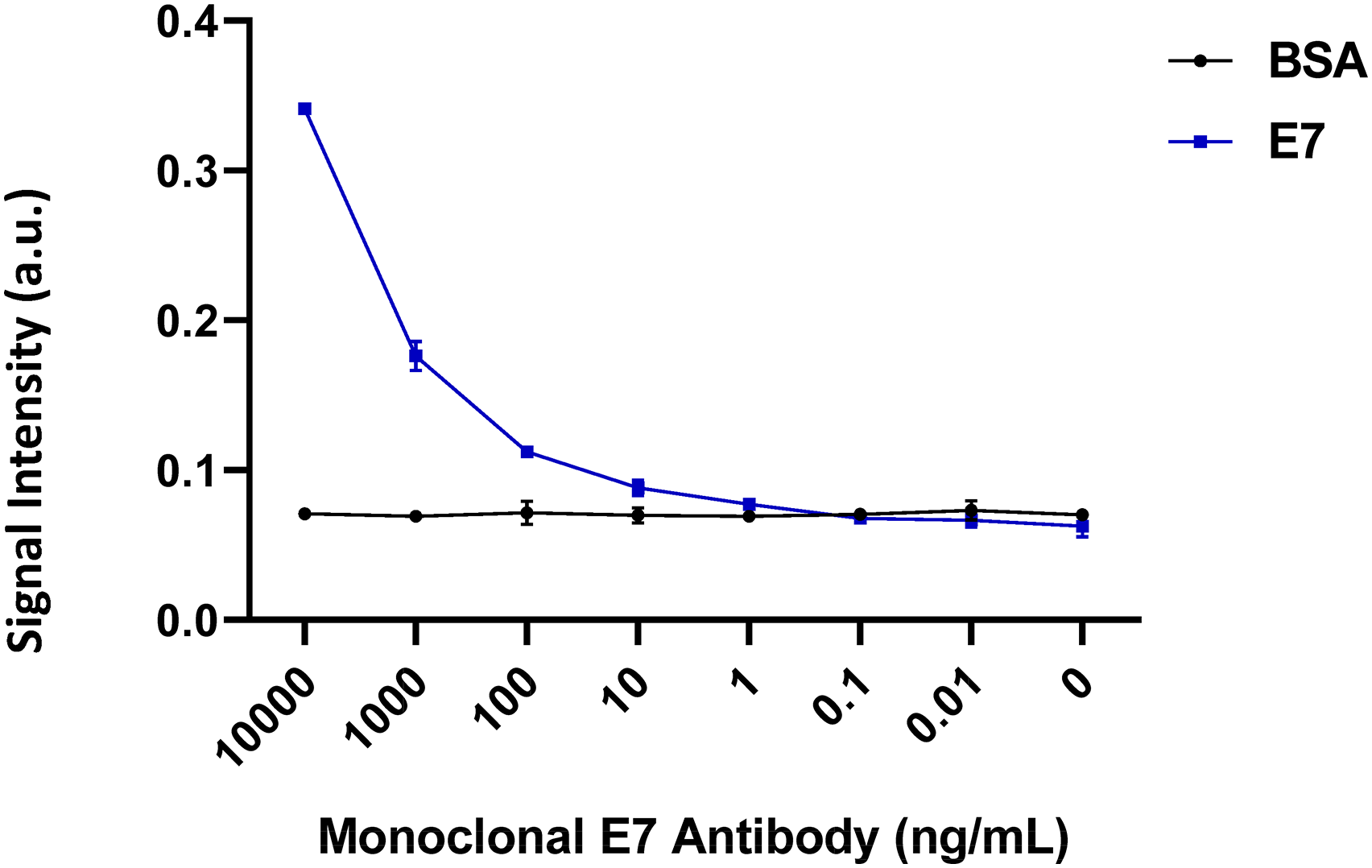
Limit of detection (LOD) of the LFA. BSA (25 μg/mL), HPV16 E7 (0.5 mg/mL) and human IgG (0.4 mg/mL) proteins were dispensed on the negative control line, test line, and positive control line. LFAs were performed using serial dilutions of a monoclonal HPV16 E7 antibody spiked in negative plasma. Each dilution was tested in triplicate. Signal intensity was calculated from the slope of the voltage -time profile using the point-of-care fluorescent reader. LOD was obtained by comparing the signal intensity of the negative control line (BSA) and test line (HPV16 E7). Error bars represent 95% confidence intervals.
Intra- and inter-assay variability for each antigen was measured and ranged from 1.5%–29.4% (Figure S3 and Supplementary Table 2). The results from the strips stored at 4 °C and room temperature for three weeks were similar. 27 Therefore, we focus on investigating the strips stored at room temperature. To determine stability of storage at room temperature, HPV16 NE6, HPV16 CE2, and HPV16 E7 were dispensed on the strips as well as a BSA negative control line and IgG positive control line. Replicate strips were sealed with desiccants under vacuum and stored at room temperature (25 °C). Assays were run monthly for 4 months. A decrease in NE6 stability, but not CE2 or E7 was observed after three months (Figure S4).
HPV16 antibody prevalence
IgG Abs to the three HPV16 antigens (CE2, NE6 and E7) were measured by the LFA in serum from 119 HPVOPC cases (collected at diagnosis), 41 partners, and compared to 81 healthy volunteers (Figure 4). The median of all HPV16 Abs were higher in cases compared to volunteers (p < 0.001). There was no significant difference observed between healthy volunteers and partners of HPVOPC cases for any HPV16 antibody except HPV16 NE6. The median in healthy volunteers (1.178) is higher than partners (1.178 vs 1.1, p = 0.0275).
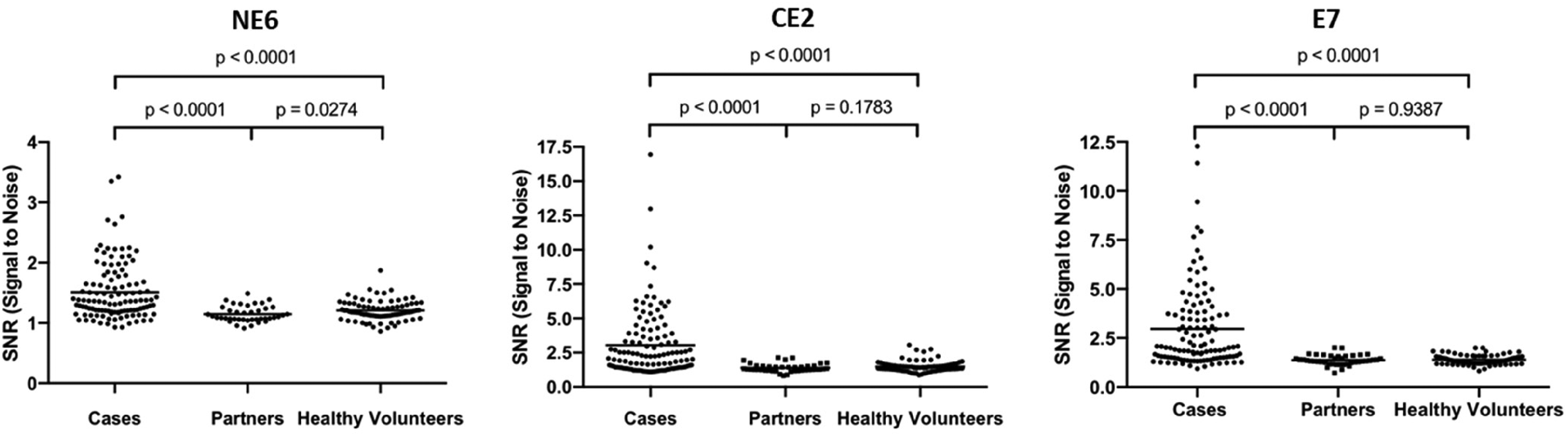
Detection of HPV16 antibodies in serum from 119 HPVOPC cases, 41 partners, and 81 controls. Human IgG specific for HPV16 NE6, HPV16 CE2, and HPV16 E7 were measured using the LFA. Signal intensity from each site was calculated from the slope of the voltage -time profile using a fluorescent reader. The signal to noise ratio (SNR) of IgG was calculated from the signal intensity of specific HPV16 protein divided by the control BSA protein in sera. Comparisons of SNR between control and case group were performed using Mann-Whitney nonparametric analysis.
Abs to E7 (68/119, 57.1%) had the highest prevalence in cases followed by CE2 (57/119, 47.9%) and NE6 (38/119, 31.9%). The majority of HPVOPC cases had Abs to at least one early antigen (CE2 NE6 and/or E7) (91/119, 76.5%). The prevalence of serum IgG Abs among HPVOPC cases is summarized in Table 1a. This data was also compared with published data using the laboratory-based RAPID ELISA with the same HPV + OPC cases and controls. 8 The sensitivity for each test was summarized in Table 1b. The positive predictive value (PPV) and negative predictive value (NPV) for this LFA platform were 93.8% and 72.8%, respectively.
HPV16 antibody prevalence.
(A) prevalence of a positive antibody response to each HPV16 protein.
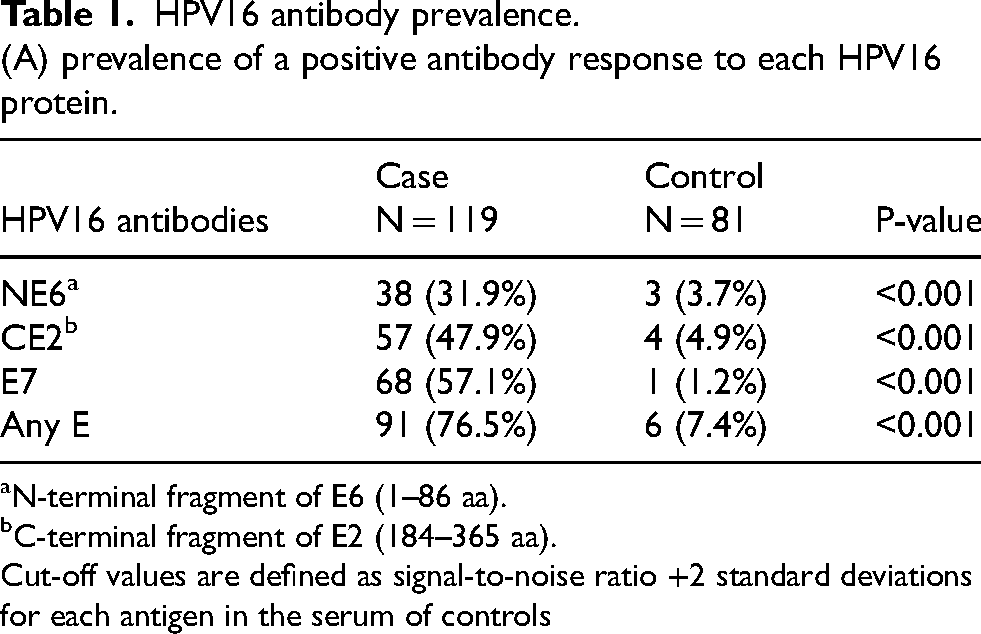
N-terminal fragment of E6 (1–86 aa).
C-terminal fragment of E2 (184–365 aa).
Cut-off values are defined as signal-to-noise ratio +2 standard deviations for each antigen in the serum of controls
(B) Comparison of LFA vs. laboratory ELISA.
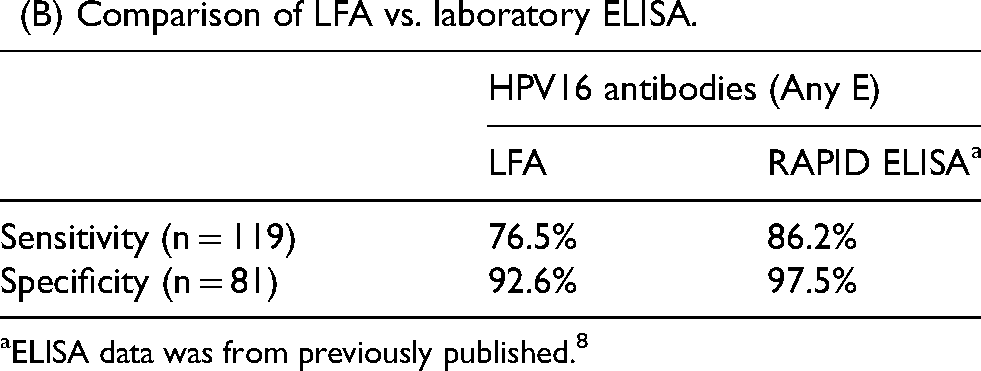
ELISA data was from previously published. 8
We calculated the receiver operating characteristic (ROC) curves for each biomarker and for the combined 3-Ab panel (Figure 5 and Table 2). The area under the curve (AUC) for each biomarker were: HPV17 E7 (AUC = 0.83), HPV16 CE2 (AUC = 0.77), and HPV16 NE6 (AUC = 0.71). At a fixed specificity of 90%, the sensitivity of discrimination of HPVOPC from healthy controls are 0.63 for HPV16 E7, 0.55 for HPV16 CE2 and 0.42 for HPV16 NE6. For the 3-Ab combination panel, the AUC was 0.86, and the sensitivity at 90% specificity increased to 0.71.
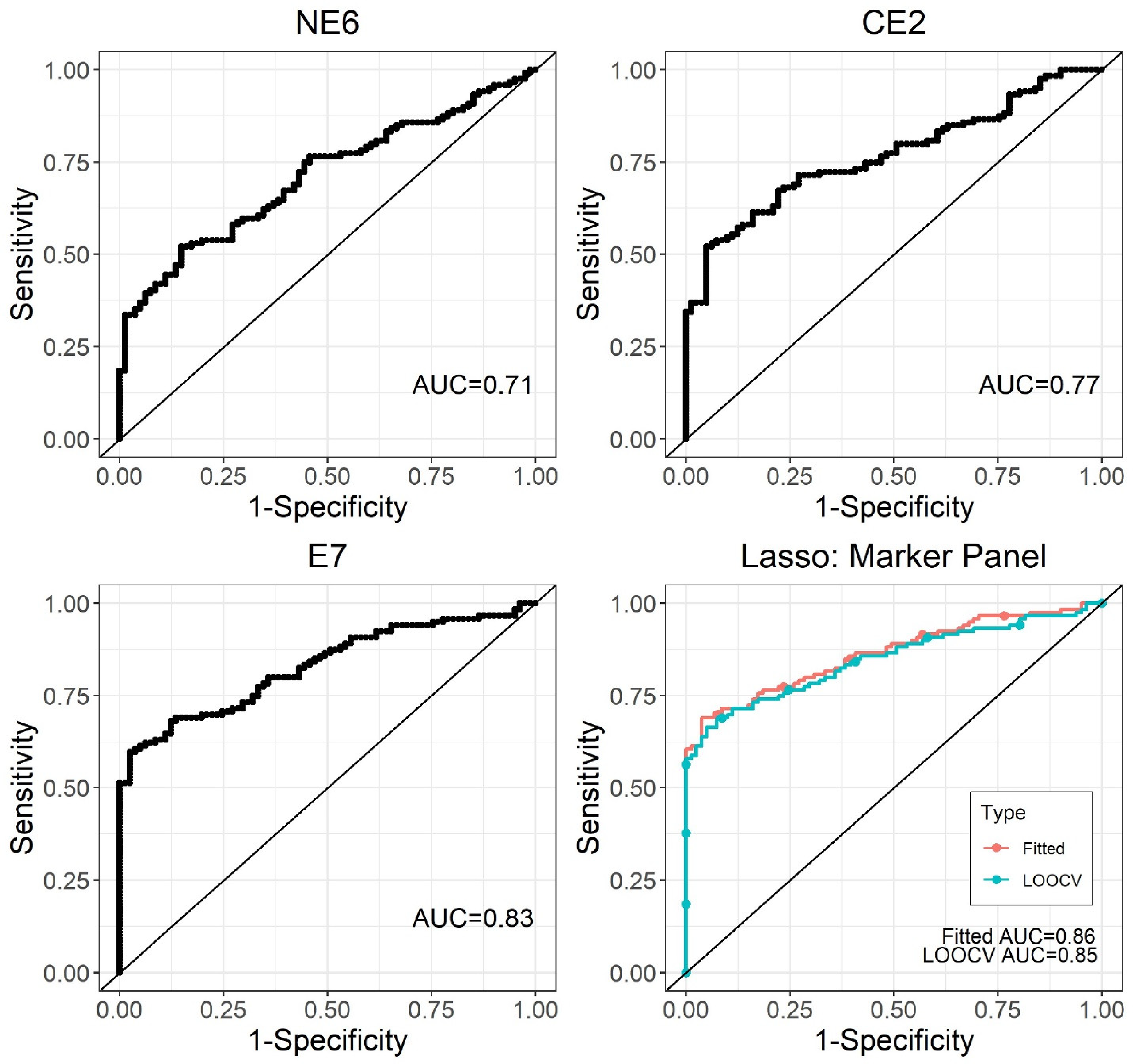
Receiver operating characteristic curves of HPV16 serologic biomarkers. Individual biomarker performance (IgG specific for NE6, CE2, E7) is shown in panels (A), (B), and (C), as well as the combined results are shown in panel (D). The ROC curve for the 3-panel HPV IgG biomarkers showed a fitted AUC of 0.86 (0.81–0.91, 95% confidence interval) with sensitivity = 0.71 at specificity = 0.9 (leave-one-out cross-validation AUC = 0.85) using Lasso regression.
Receiver operating characteristics of each HPV16 biomarker.
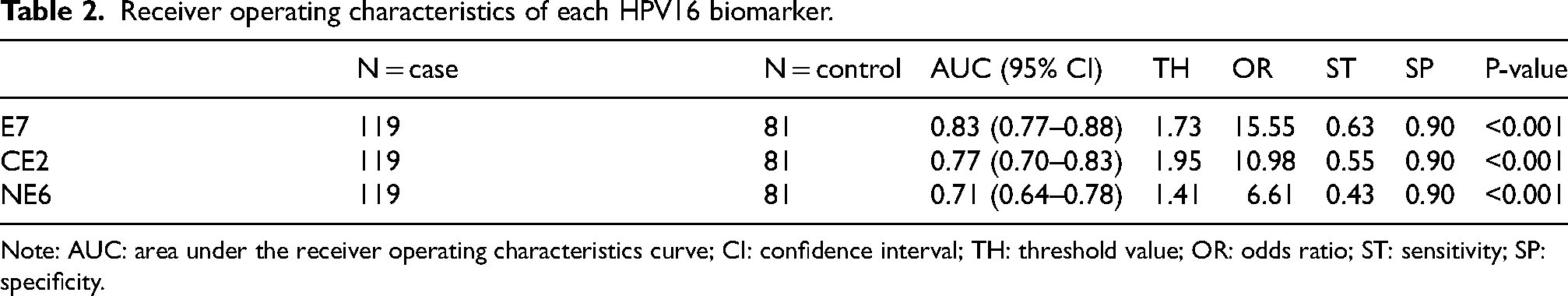
Note: AUC: area under the receiver operating characteristics curve; CI: confidence interval; TH: threshold value; OR: odds ratio; ST: sensitivity; SP: specificity.
Discussion
In this study, we developed a rapid, inexpensive, and user-friendly fluorescent serological LFA for HPVOPC in a multiplex format. The assay requires less than 1 µL of plasma/serum to test three HPV antigens: HPV16 CE2, HPV16 NE6, and HPV16 E7 simultaneously. These three antigens were selected because circulating antibodies against HPV16 E6, HPV16 E2, and HPV16 E7 proteins are highly specific biomarkers for HPVOPC,12,28,29 and they are detectable years before clinical diagnosis, offering a possible utility for early detection and screening.10,12,13,30 HPV16-specific antibodies, such as HPV16 E2, HPV16 E1, and HPV16 E6, have been associated with improved overall and progression-free survival in patients with HPV-associated oropharyngeal carcinoma. 31 However, their utility for screening remains unclear, as there is currently no therapeutic intervention to offer anyone identified with these antibodies who does not have cancer upon examination. Despite this, there is significant potential for their use in light of the current lack of screening tools and increasing rates of HPVOPC.
Overall, the combined three-proteins assay displayed a sensitivity of 76.5% and specificity of 92.6% for HPVOPC detection. While the ELISA data of HPV16 NE6 was not as promising as the other two antigens (CE2 and E7), it showed observable differences between the case and control only at the first dilution point (Figure 2(d)). However, HPV16 NE6 still produced reliable results using a larger sample set: HOTSPOT (Figure 4). Antibodies to E7 (57.1%) were the most prevalent, followed by CE2 (47.9%) and NE6 (31.9%) proteins (Table 1). However, unlike other studies where HPV16 E6 seropositivity emerges as a highly reliable single diagnostic marker for HPV16-driven HNSCC,12,29,32 this LFA assay has lower sensitivity for detection of antibodies to E2 and E6 antigens. This discrepancy could be attributed to the use of protein fragments to improve yield and stability in our LFA assay (Supplementary Table 1). Other groups have been developing point-of-care approaches for HPV serology, but challenges remain, such as limited multiplexing 33 and long assay times. 34
Reproducibility and quantitation have been ongoing challenges for LFAs, especially in serological testing with fingerstick samples.35,36 We evaluated intra- and inter-assay reproducibility, aiming for coefficients of variation under 20% (Figure S3 and Supplementary Table 3). To mimic long-term storage at room temperature, we sealed the strips in replicate bags with desiccants under vacuum. Over time (Figure S4), the signal of anti-HPV16 NE6 antibody decreased by 30% after 3 months of storage, while HPV16 CE2 and HPV16 E7 specific antibodies remained stable. The strips were found to be stable for at least two months of storage at room temperature. While this is not as long-lasting as FDA-authorized at-home LFA with shelf-lives of about 4 months to two years from the day of manufacture, 37 the storage is likely to improve significantly with highly disseminated optimization techniques.38–40
In our study, the LOD of our assay for detection of HPV16 E7 in serum is about 10 ng/mL (Figure 3) which is comparable to other LFA platforms. For future developments, we can explore and optimize the detection techniques and nanoparticles to further improve the LOD. For example, the LOD for the detection of IgG against SARS-CoV-2 ranges from 5 ng/mL to 20 ng/mL.41,42 In order to enhances sensitivity, surface-enhanced Raman spectroscopy (SERS) has been combined with LFAs, resulting in an improved LOD for IgG against SARS-CoV-2, reaching 1 pg/ mL. 43
As expected, the analytic performance of the LFA was lower than our laboratory-based RAPID ELISA (Table 1B, sensitivity 76.5% vs. 86.2%), which was attributed to the use of chemiluminescence for signal amplification in the laboratory assay. LFAs inherently have short times for antibody-antigen interactions, with brief (seconds) contact compared to the one-hour incubation time in RAPID ELISA. Additionally, RAPID ELISA involved more rigorous washing steps than the LFA, which helps reduce background signal. More, this LFA utilized bacterial production of proteins to achieve large-scale testing yields, which involved using fragments of E2 and E6 proteins, limiting the available antigenic determinants.
The primary target population for this assay includes high-risk individuals, such as men over the age of 50 or those with a history of exposure to high-risk HPV infection. Given the low prevalence of HPVOPC in general populations, secondary assays will be needed for confirmation. We observed a prevalence of antibodies to E protein in controls (7.4%) (Table 1A), which is similar to the prevalence of the HPV16 DNA test (10%). The relative clinical utility and timing of HPV nucleic acid testing versus serology remains to be determined in prospective clinical trials. Lateral flow serology offers specific cost and logistic advantages as a primary screening tool, while laboratory-based serology assays, with higher specificity, 8 could serve for confirmation.
HPV serologic testing offers a potentially promising approach for the early detection of HPVOPC among higher risk groups to identify individuals with high risk of disease. However, it is currently unclear how to best follow or manage those identified in a serologic HPV16 screen. Few studies have directly compared the different HPV biomarker classes in oral rinses and serology. One study compared biomarker performance in 133 incident HPVOPC and 134 noncancer controls, evaluating the sensitivity and specificity of HPV DNA (DEIA and Cobas), RNA (Aptima), and serological detection. 28 In that study, the sensitivity of oral HPV DNA/RNA detection was relatively low, with DEIA at 51%, Cobas at 43%, and Aptima at 23%. In contrast, the sensitivity of HPV16 E6 antibody detection was 88%. Importantly, all these tests demonstrated high specificity for HPVOPC (>98%), although that varies by detection method. 28 In healthy individuals oral HPV infections are usually self-cleared,28,29,34,44 with a median time to clearance of 1.4 years,45–48 during which HPV DNA may only be detected transiently. In contrast, HPV antibodies persist, making them easier to detect. In the longitudinal HOUSTON screening study of middle-aged men, oral HPV nucleic acid and serology tests were conducted simultaneously, and both biomarkers were associated with detection of HPV-associated disease. 24
While there are currently no FDA-approved commercial assays for HPV head and neck cancer screening, multiple comparable products are available for cervical cancer screening, such as the Cepheid GeneXpert, Roche Cobas testing, and Aptima HPV testing.49–51 Research indicates that women harboring persistent HPV-16 infection in the cervix for less than one year face a 40% risk of developing cervical intraepithelial neoplasia grade 2 or higher within three years. 52 However, the natural progression of HPV infection in the oropharynx, from initial infection to carcinogenesis, remains largely unknown, leaving numerous questions unanswered. Despite efforts to evaluate the prevalence of HPV-16 DNA in saliva through various studies,53–56 clinical assessments of positive individuals have not been consistently conducted. Moreover, studies focused on clinically assessing individuals with persistent HPV infection for premalignancy or microscopic carcinoma have yielded inconclusive results.56,57 Consequently, there is a prevailing view that DNA screening for early occult or premalignant oropharyngeal lesions may not be feasible. Therefore, exploring HPV16 protein serology holds promise for clinical investigations into HPVOPC screening. 11
According to WHO data, the cost of existing HPV DNA/mRNA tests for cervical cancer, such as the Cepheid GeneXpert, Roche Cobas, and Aptima HPV tests, ranges from $7.90 to $14.90 per reagent, excluding instrument and maintenance costs. Including sample collection and other associated expenses, these prices can increase by $1 to $6. 58 In comparison, lateral flow assays such as at-home COVID tests have reagents costs as low as $2–$559–61 with bulk purchasing and high-volume, low-cost manufacturing. LFA platforms present a cost-effective alternative for population-based screening.
There remain limitations in the current format of the LFA assay, including the sensitivity and instability of HPV16 E6 with storage at room temperature, as well as the fluorescent microspheres that require sonication before use. However, we focused our efforts on limiting the cost of materials, equipment, time, and the need for minimal operator training, to be compatible with global health and resource-constrained applications. Given the success of screening tools for cervical cancer and ∼100,000 newly diagnosed cases of OPC annually, 62 the demonstration of this LFA provides compelling evidence for the potential of a highly deployable, low-cost assay to aid in the urgent need for early detection to improve patient outcomes and reduce the ∼48,000 annual deaths.
Supplemental Material
sj-docx-1-cbm-10.1177_18758592241311183 - Supplemental material for Development of a multiplexed lateral flow assay for the serologic detection of HPV-associated head and neck cancer
Supplemental material, sj-docx-1-cbm-10.1177_18758592241311183 for Development of a multiplexed lateral flow assay for the serologic detection of HPV-associated head and neck cancer by Ching-Wen Hou, Pankaj Kumar, Stacy Williams, Meilin Zhu, Uwa Obahiagbon, Joshua Eger, Gypsyamber D'Souza, Yunro Chung, Lalit Dar, Neerja Bhatla, Jennifer Blain Christen and Karen S Anderson in Cancer Biomarkers
Footnotes
Acknowledgments
We would like to thank laboratory members Meredith Smith, Emma Johnson, Rachel Fisher, Ntombizodwa Makuyana, Nicholas Shein, and Evelyn Ladipo for protein production. We thank Dr Roberta Reed for critical review of the manuscript. This study was supported by the National Cancer Institute Cancer Detection, Diagnosis, and Treatment Technologies for Global Health grant NCI CA211415: Rapid Point-of-Care Detection of HPV-associated Malignancies.
ORCID iDs
Author contributions
Conception: CWH, PK, MZ, UO, JE, GD, LD, NB, JBC, and KSA. Interpretation or analysis of data: CWH, PK, SW, and YC. Preparation of the manuscript: CWH, SW, YC, and KSA. Revision for important intellectual content: PK, SW, YC, JBC, and KSA. Supervision: LD, NB, JBC, and KSA.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded through grant support from the National Cancer Institute Cancer Detection, Diagnosis, and Treatment Technologies for Global Health grant NCI CA211415: Rapid Point-of-Care Detection of HPV-associated Malignancies.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Drs. Anderson and Blain Christen are founders of FlexBioTech, a point of care diagnostic company.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.