
Abstract
Objective
In NSCLC, the main approach to differentiate between intrapulmonary metastases (IPM) and multiple primary lung cancer (MPLC) is to integrate histopathological and genomic information. Here, we identified viable biomarkers that can distinguish IPM from MPLC by integrating comprehensive genomic profiling (CGP) and targeted RNA sequencing.
Methods
We retrospectively collected tissues from at least two lesions in 34 patients. 29 and 5 out of 34 patients determined as pathologic MPLC (pMPLC) and pathologic IPM, respectively, according to Martini-Melamed criteria (M-M criteria). A comprehensive investigation at genomic and transcriptomic level was conducted.
Results
Nine of the 29 pMPLCs shared trunk mutations in their lesions and were consequently reclassified as IPM. Survival analyses revealed that classification integrated M-M criteria and mutational profiling could distinguish IPM/MPLC more accurately. Further exploration at the transcriptomic level revealed elevated expression levels of genes related to epithelial-mesenchymal transition and immunomodulatory pathways in IPM. Notably, the expression of CXCL1 and CXCL8 was significantly upregulated in IPM.
Conclusions
We found that the expression of CXCL1 and CXCL8 in any tumor lesion within a patient could reliably indicate IPM. Additionally, assessing the transcriptional levels of CXCL1 and CXCL8 also provide a dependable and practical approach to identify IPM from MPLC.
Keywords
Introduction
Lung cancer stands as the leading cause of cancer-related fatalities on a global scale. The advent of early screening methods, particularly low-dose computed tomography (LDCT), has facilitated the increased detection of multiple pulmonary lesions associated with lung cancer. 1 These lesions, classified based on their origin, fall predominantly into two categories: multiple primary lung cancer (MPLC) and intrapulmonary metastasis (IPM)-related lung cancer. In contrast to IPM, MPLC refers to the occurrence of two or more primary lung cancers in different lobes or segments within one or both lungs of the same patient. Depending on the timing of their appearance, MPLC is further categorized as synchronous MPLC (sMPLC) or metachronous MPLC (mMPLC).2,3 The ability to distinguish between MPLC and IPM carries significant implications for tumour staging, treatment strategy, and prognosis. This differentiation is vital for tailoring precise and personalized treatments for lung cancer patients. 1
The Martini-Melamed (M-M) criteria, introduced in 1975, were the pioneering guidelines for distinguishing between MPLC and IPM. These criteria utilized parameters such as tumor location, pathological morphology, and the presence of distant metastases to differentiate MPLC. Despite remaining the most widely adopted diagnostic framework in clinical practice, it is noteworthy that patients diagnosed with primary lung cancer (stages I and II) according to the M-M criteria might actually have underdiagnosed lung metastases (T4 or M1a). Consequently, previous reports have indicated varying 5-year survival rates for patients with sMPLC, ranging from 34% to 45%. 4
Subsequently, several diagnostic criteria for MPLC have been proposed and subsequently revised multiple times in clinical practice. For example, the 2013 American College of Chest Physicians (ACCP) guidelines 5 represent one such effort. Nevertheless, it's worth noting that the ACCP guidelines acknowledge that many diagnostic criteria for MPLC can only serve as references. A comprehensive analysis of clinical manifestations and radiographic characteristics remains essential. This underscores the absence of a universally accepted gold standard, emphasizing that the accuracy of MPLC diagnosis depends on the input of experts from various fields, particularly radiologists and pathologists.
As gene research methods and technologies have progressed, several molecular techniques, including array comparative genomic hybridization (aCGH), 6 microsatellite analysis (MSA), 7 Next-Generation Sequencing Technology (NGS),8,9 and whole-exome sequencing (WES),10,11 have been employed to profile the genetic attributes of MPLC.
Presently, genetic testing primarily focuses on analyzing specific aspects of DNA. However, variations in the number of genes incorporated into different tests, coupled with significant disparities in sensitivity and specificity, continue to introduce diagnostic biases. In this study, we undertake a comprehensive and systematic evaluation of the heterogeneity between MPLC and IPM. We achieve this through a combination of genomic and transcriptomic analyses, encompassing tumor mutations, activation status of tumor cell signaling pathways, and the immune microenvironment. This approach aims to provide evidence for a deeper comprehension of MPLC, the distinction between MPLC and IPM, and ultimately, the identification of unique molecular biomarkers.
Materials and methods
Patients and samples
Between February 2017 and March 2019, a total of 34 patients, aged over 18 years, with an ECOG score of 0-1, who had not undergone neoadjuvant therapy and had undergone surgical resection for ipsilateral simultaneous multifocal lung cancer, were recruited for this study based on the M-M criteria. We retrospectively collected tissues from at least 2 lesions from each patient at the Affiliated Hospital of Qingdao University, situated in Shandong Province, China.
Clinical information
We gathered demographic and clinical data from patients, which included age, gender, smoking history, the number of lesions, histology, histological subtype, surgical approach, as well as tumor location and size assessed by the pathologist. Survival status was tracked through periodic outpatient records and telephone follow-ups until March 2023. This study received approval from the Institutional Review Board of the Affiliated Hospital of Qingdao University (QYFY WZLL 28029). All participants provided written informed consent, and their clinical records were obtained.
DNA and RNA extraction, library preparation
Following protocols outlined in previous literature12,13 with slight modifications, we extracted DNA and RNA from fresh cytological specimens using the AmoyDx Tissue DNA/RNA Kit (AmoyDx Inc, Xiamen, China). Quantification of DNA and RNA concentration was performed using the Quantus fluorometer, Quantus dsDNA (Cat. # E2670 Promega), and RNA HS Assay Kit (Cat. # E3310, Promega). Fragment length was assessed using the Agilent 2100 Bioanalyzer. We prepared NGS libraries for DNA fragments and strand-specific DNA libraries after RNA reverse transcription, respectively (Cat. #E7645/#E7760L, NEB).
Capture and sequencing
The DNA and RNA libraries were captured using the AmoyDx® Master Panel, which contains 571 genes for DNA mutation (single-nucleotide variation [SNV], insertion/deletion [Indel], fusion, copy number variation [CNV], microsatellite instability and tumor mutation burden [TMB]) detection and 2660 genes for RNA expression and fusion detection. The DNA samples were sheared into 200–250 bp fragments using Covaris LE220 (Woburn, MA, USA) and indexed NGS libraries were then prepared by performing end-repair, A-tailing, adaptor ligation, and amplification procedures, using the NEBNext® Ultra™ II DNA Prep Kit (Cat. #E7645, NEB). The RNA samples were fragmented at 95 °C for a duration determined by the DV200 value estimated using the Agilent 2100 Bioanalyzer System. The fragmented RNA then underwent reverse transcription, complementary RNA synthesis, and strand-specific library preparation using the NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina® (Cat. #E7760L, NEB). After pooling and qualifying the library, sequencing was performed on the Illumina NovaSeq 6000 instrument (Illumina). The results underwent laboratory quality control, analysis, and annotation. For processing sequencing data, quality assessment was carried out using an in-house script, FormatFastQ (v2.4.0). Alignment to targeted genes from Gencode hg37 was performed using STAR (v2.7.2b), and gene counts were quantified using RSEM (v1.3.3). Sequencing data were initially cleaned to remove sequencing adaptors and low-quality reads (quality < 15) or poly-N with Trimmomatic. These reads were then mapped to the human reference genome, version 19 (hg19), using the Burrows-Wheeler Aligner. Annotation of the vcf files was conducted using analysis of variance. SNVs and Indels that were called underwent further filtering, requiring a minimum of ≥ 5 variant-supporting reads, ≥ 5% variant allele frequency supporting the variant, > 2% population frequency in the 1000 g or ExAC or GnomAD database, location in the CDS region, and absence of annotation as (likely/predicted) oncogenic in the OncoKB database. These filtered variants were considered functional and used for subsequent data analysis.
Validation dataset
For validation purpose, the clinical data and RNA sequencing results of 32 NSCLC patients admitted to this hospital earlier were reviewed and sorted out. The clinical information of these patients is shown in supplementary Figure 1A.
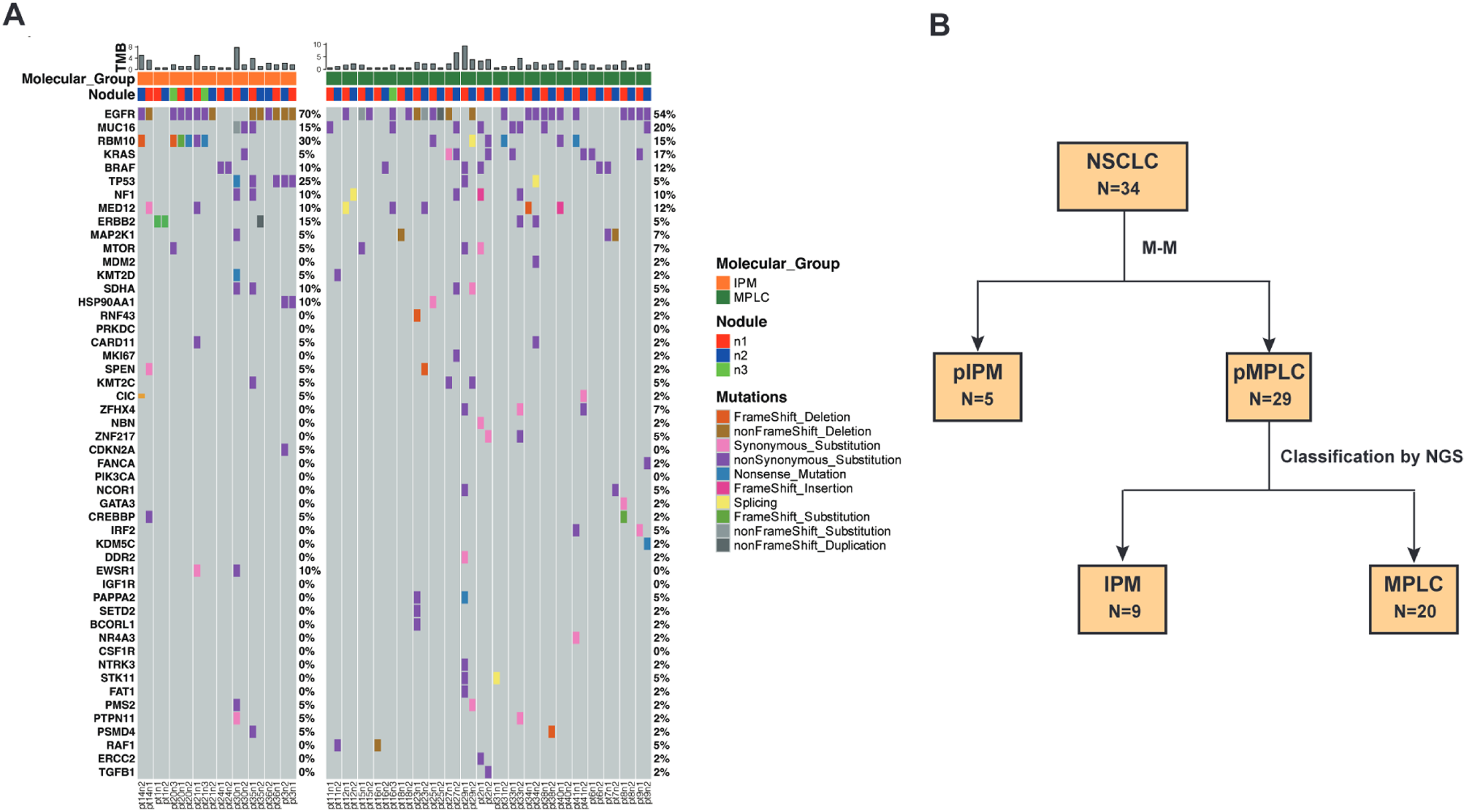
Molecular analysis of the 29 pMPLC patients. (A) Waterfall Plot illustrates the distinct driver gene mutation profiles observed in the current molecular classification, which encompasses IPM and MPLC, within the context of pathologic multiple primary lung cancer (N = 29). (B) The integration classification process flowchart.
Real-time quantitative reverse transcription-polymerase chain reaction (qrt-PCR) analysis of CXCL1 and CXCL8 mRNA expression
Expression data validation was performed by qRT–PCR using RNA extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissues with TRIzol Reagent (Thermo Fisher Scientific Inc.). RNA was converted to cDNA using the HiScript® II Q Select RT SuperMix for qPCR (R232-01; Vazyme) according to the manufacturer's instructions (55°C for 15 min and 85°C for 15 min). qRT-PCR was carried out using the SYBR Green qPCR Master Mix (Q712-02; Vazyme) according to the manufacturers’ instructions by an ABI 7500 real-time PCR system (Thermo Fisher Scientific Inc.) and amplified with target gene specific primers. The forward and reverse primers for CXCL1 were 5′-AAGTCATAGCCACACTCAAG-3′ and 5′-TCAGTTGGATTTGTCACTGT-3′; and the forward and reverse primers for CXCL8 were 5′-GAGAGTGATTGAGAGTGGAC-3′ and 5′-GAATTCTCAGCCCTCTTCAA-3′; and the forward and reverse primers for β-actin were 5′-GACATTAAGGAGAAGCTGTG-3′ and 5′-CATGATGGAGTTGAAGGTAG-3′, respectively. The reaction conditions included an initial step at 95°C for 3 min, and 42 cycles at 95 °C for 10 s and at 60 °C for 3 min, 72 °C for 1 min and final extension at 72 °C for 10 min. All experiments were performed in triplicate with three technical replicates.
Statistical analysis
All statistical analyses were conducted using the rms package in the R language (version 4.2.1). Demographic characteristics and the mutational landscape of patients were examined through descriptive statistics. Significance regarding the correlation between clinicopathological parameters and gene mutation status was determined using Pearson's chi-square test for categorical variables and Fisher's exact test. All tests were two-sided, and differences were considered significant at p < 0.05.
Results
Clinicopathological characteristics of patients
A total of 34 patients were included in this study, with an average age of 60.47 years (range, 29 to 77 years). The number of female patients (n = 26, 76.47%) significantly exceeded that of male patients (n = 8, 23.53%). Among the patients, 28 (82.35%) had no history of smoking, while 6 (17.60%) had a smoking history. All patients underwent initial surgical resection without receiving neoadjuvant therapy. Among them, 3 patients (8.82%) presented with 3 surgical lung lesions, while the remaining 31 patients (91.18%) had 2 surgical lung lesions. Of these, 20 patients (58.82%) had lesions located in the same lobe, and 14 patients (41.18%) had lesions located in different lobes of the ipsilateral lung. All of these cases underwent NGS sequencing (Table 1). Surgical procedures mainly included video-assisted thoracoscopic surgery (VATS, 72.41%) and robot-assisted thoracoscopic surgery (RATS, 27.59%). The types of surgery performed included lobectomy in 17 cases (58.62%) and sublobectomy in 11 cases (37.93%), with one case having unspecified surgical methods (as shown in Table 2).
Postoperative pathology revealed differences in multiple pulmonary lesions (Table 3). The majority of lesions in the pMPLC group were located in the right upper lobe (RUL, 37.70%), followed by the left upper lobe (LUL) at 14.75%, right lower lobe (RLL) at 43%, right middle lobe (RML) at 8.2%, and left lower lobe (LLL) at 9.84%. The pMPLC group showed fewer solid lesions (SN) and more pure ground-glass opacities (pGGO) lesions and mixed ground-glass opacities (mGGO). The mean tumor size was 1.35 ± 0.88 mm, with invasive adenocarcinoma (IA) infiltration in 48 cases (78.69%) and minimally invasive adenocarcinoma (MIA) in 13 cases (21.31%).
Efficacy of molecular classification to distinguish IPM from MPLC
Based on clinicopathological features, among the 34 patients, 29 patients were found to fit the M-M criteria for MPLC (pMPLC), and the remaining 5 patients were IPM (pIPM). The average age of the 29 pMPLC patients was 60.90 years (range, 29 to 77 years), and females accounted for 72.41% with a majority having no history of smoking (79.31%). We detected 76 lesions from 34 patients through NGS testing with a Master Panel (570 DNA genes and 2661 RNA genes), and mutational profiling was used to evaluate the clonal relatedness of tumors. Interestingly, among the 29 pMPLCs, 20 patients (74.07%) had tumors that did not share a common mutation, and were clearly considered to be of differing clonal origin, therefore, according to previous report, 14 these patients were classified as MPLC. Nine of the 29 pMPLC patients exhibited the same mutated genes in different lung lesions and should be classified as IPM (Table 4). Differences in the mutation profiles of the driver genes in IPM and the remaining MPLC are demonstrated in Figure 1A. Alterations in the EGFR, MUC16, RBM10, KRAS, and BRAF genes were the most frequently observed mutations, occurring in 63.64%, 20.78%, 18.18%, 12.99%, and 11.69% of cases, respectively. The classification process flowchart showed the IPM/MPLC subtypes of the 34 patients determined by integrating M-M criteria and molecular assessment (Figure 1B). Furthermore, to explore the integration classifier prediction efficacy of survival outcomes in patients, survival analyses were performed. The median follow-up time for the patients was 58.5 months. Patients with MPLC displayed statistically significant better DFS and OS compared with the group of pIPM and IPM (P = 0.008 and 0.032, Figure 2).
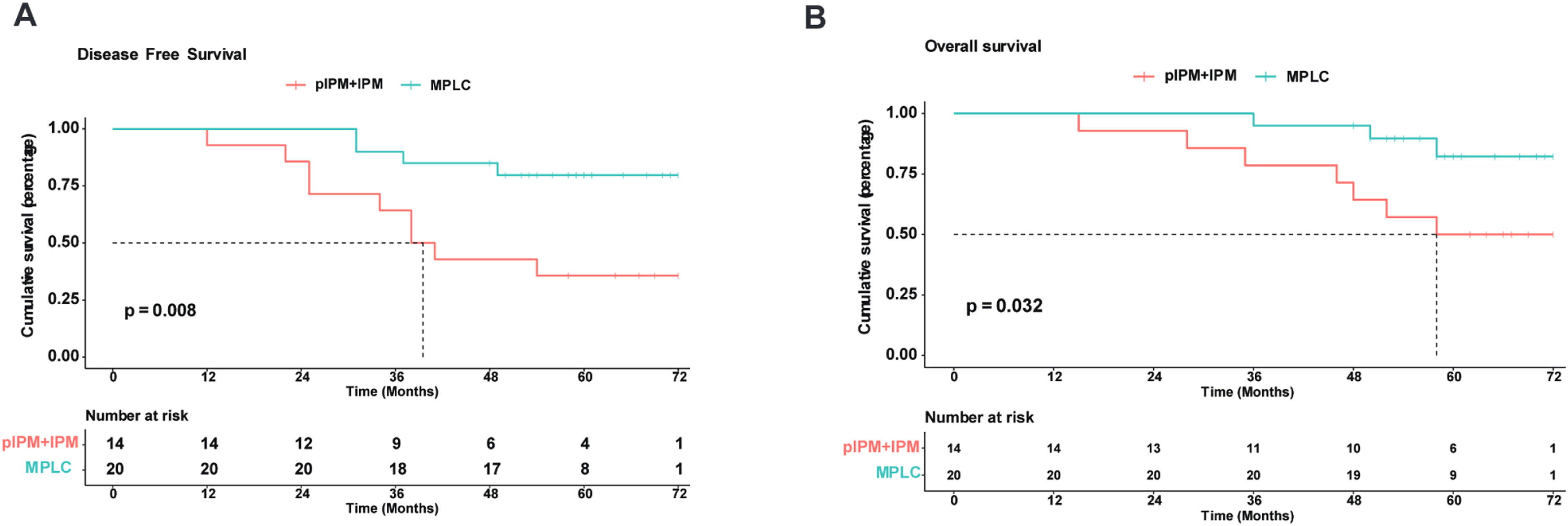
The survival (OS) and disease-free survival (DFS) rates of lung cancer patients are presented in the following figures: Panel (A) displays the OS for MPLC and IPM + pIPM, and panel (B) shows the DFS for MPLC and IPM + pIPM.
Differences in transcriptome profiles and molecular pathways between IPM and MPLC classified by molecular characterization
The transcriptome gene enrichment analysis in the two types of lung cancer patients classified by molecular characterization was conducted using a volcano plot. Significant differences in the transcriptome were observed between IPM and MPLC tissues. The volcano plot highlighted 35 genes with a fold change threshold > 2 and an adjusted p-value below 0.05 (Figure 3A). Specifically, compared with MPLC, genes such as CTSG, GSTT2, MMP7, CD24, and USP9Y exhibited decreased expression in IPM, whereas BMPR1B, ARG2, TNFSF4, CXCL8, CDK6, KLRC2, EGR3, CXCL1, CXCL2, NGF, CCL20, CXCL6, SERPINE1, KLRC1, IFNA1, IFNB1and etc. genes showed increased expression in IPM.
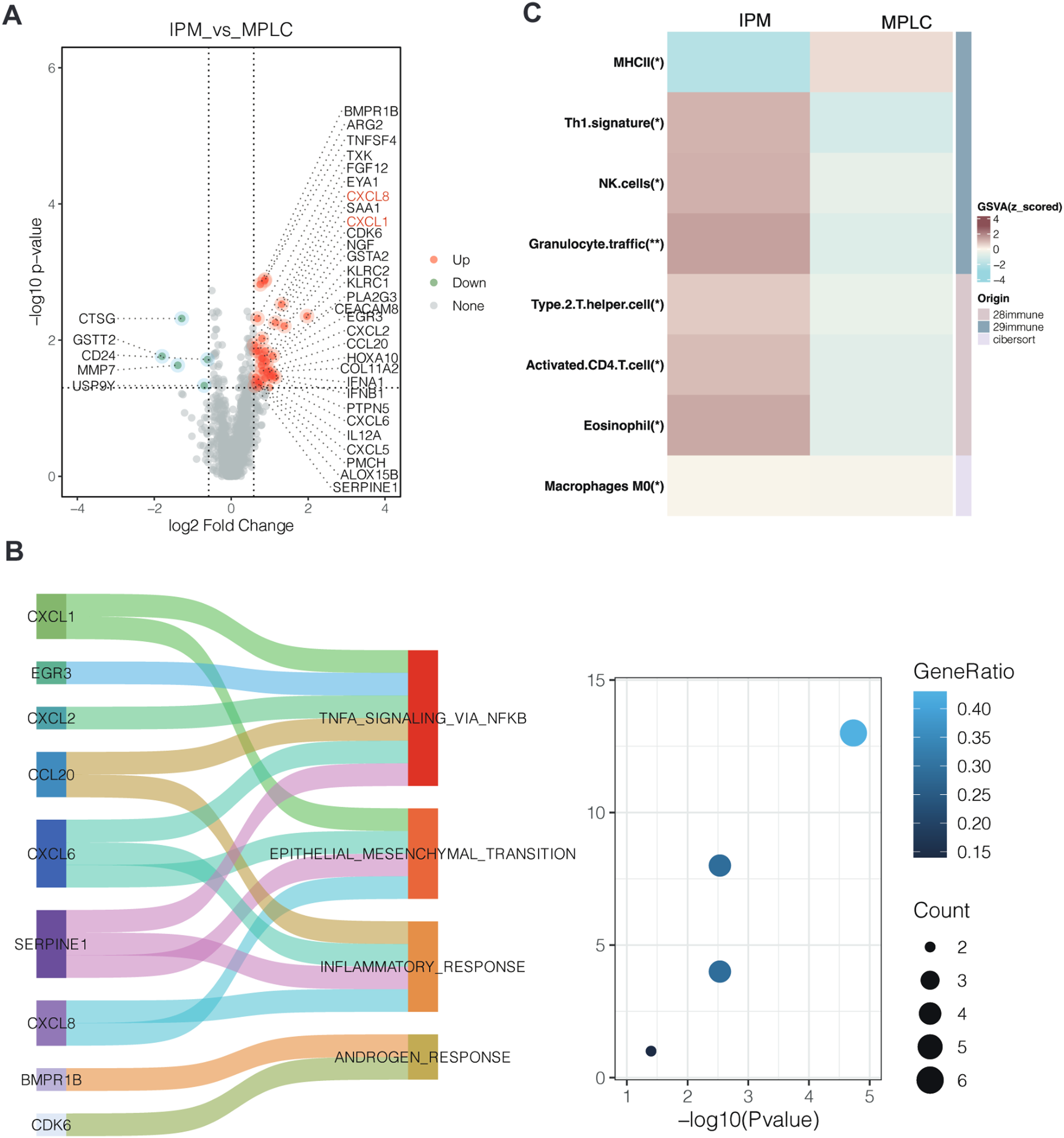
Differences in molecular characteristics between MPLC and IPM are depicted in the following panels: Panel (A) presents a volcano plot illustrating the differentially expressed genes (DEGs) between IPM and MPLC. Panel (B) showcases the results of a hallmark pathway analysis based on the DEGs identified in IPM. Panel (C) compares the distinctions in the tumor microenvironment between MPLC and IPM.
Hallmark Enriched Pathway analysis results indicated that the epithelial-mesenchymal transition (EMT) pathway and other cancer-associated pathways, including TNFA-signaling- via-NFκB, inflammatory-response, and androgen–response were enriched in IPM tissues (Figure 3B). Furthermore, using the ssGSEA method to evaluate the infiltration of 28 immune cell subtypes, 29 immune-related gene sets, and CIBERSORT analysis in the tumor microenvironment, Figure 3C showed that the infiltration levels of Th1.signature, NK.cells, Granulocyte.traffic, Type.2.T.helper.cell, Activated.CD4.T.cell, and Eosinophil in the IPM group were significantly higher than those in the MPLC group (p < 0.05), while there was no statistical significance for the other cell subtypes. Furthermore, based on the ssGSEA scores of these analyses above, the IPM group had significantly lower scores than the MPLC group for MHCII (p < 0.05) as shown through clustering analysis.
Transcriptional levels of CXCL1 and CXCL8 can assist to distinguish IPM from MPLC
Further analysis revealed significant differences in the expression of four genes, CXCL8, CXCL1, CXCL6 and SERPINE1, in the EMT pathway between IPM and MPLC patients classified on the basis of molecular features (Figure 4A). Univariate logistic regression analysis corroborated this distinction between IPM and MPLC, among which CXCL8 (OR = 1.75, CI: 1.18-2.60, P = 0.0055) and CXCL1 (OR = 1.72, CI: 1.16-2.55, P = 0.0073) were strongly correlated with IPM (Figure 4B). The expression of CXCL1 and CXCL8 in IPM and MPLC tumor tissues was compared by qRT-PCR, as shown in Figure 4C, the relative expressions of these two genes were significantly higher in IPM tissues. Further verification was conducted in the detection results of 63 samples from another 32 NSCLC patients, and it was also observed that the expression of CXCL1 in IPM was significantly higher than that of MPLC (P = 0.034), and identified a trend to higher CXCL8 expression in IPM compared to MPLC (P = 0.33, Supplementary Figure 1B). The mean area under the receiver operating characteristic curve (AUC) for CXCL8 and CXCL1 in predicting IPM were 0.729 and 0.714, respectively (Figure 4D). To assess the stability of CXCL8, and CXCL1 in classifying IPM/MPLC, a computer simulation was conducted. In this model, a random lesion sample was chosen from each patient, and OR values were calculated, repeated 1000 times. The average AUC value was approximately 0.71, indicating that CXCL8 and CXCL1 could serve as crucial and stable biomarkers for distinguishing IPM from MPLC (Figure 4 E-G).
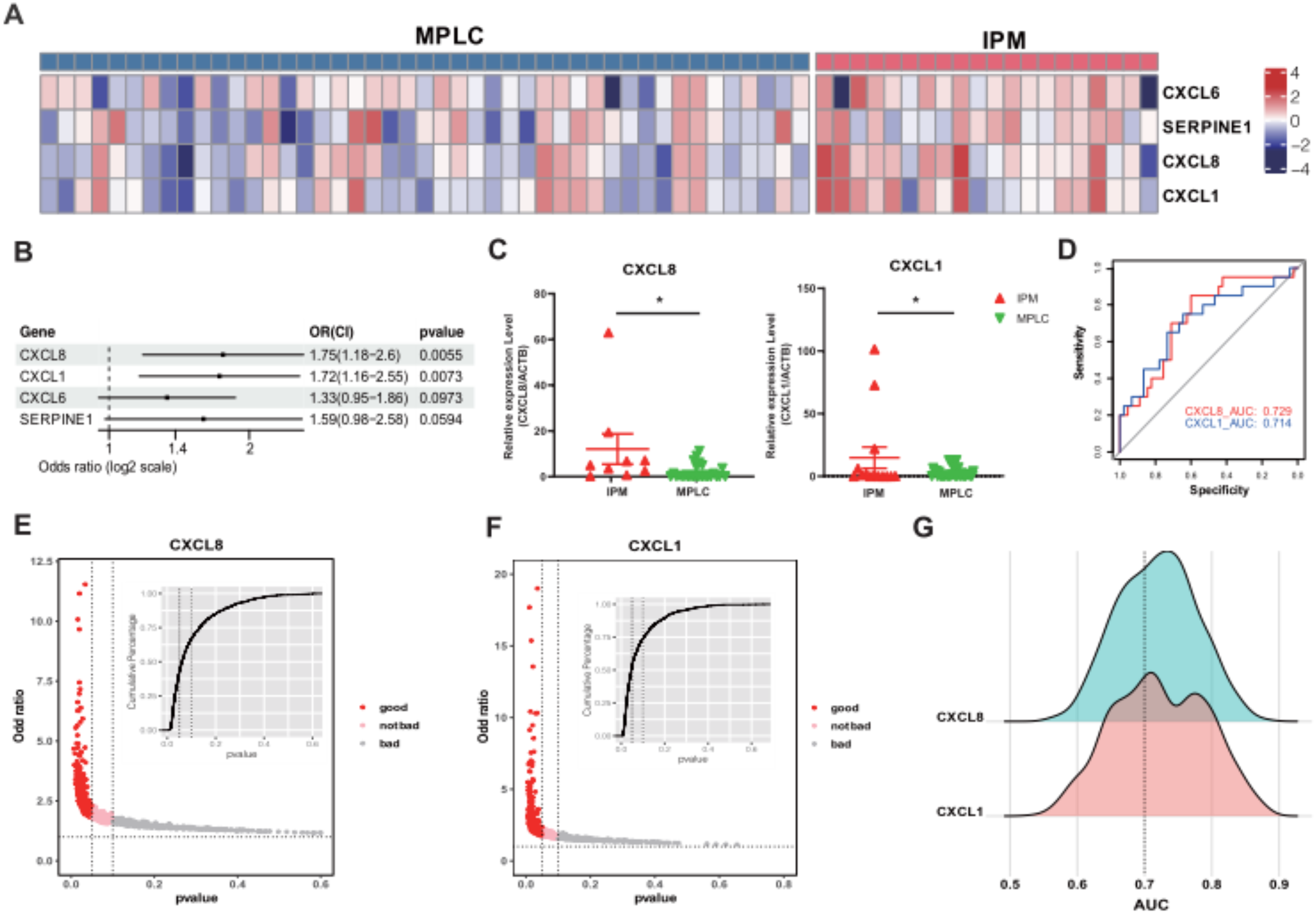
Differences in transcriptome characteristics related to epithelial-mesenchymal transition (EMT) between MPLC and IPM. Panel (A) presents a heatmap displaying the expression of CXCL8, CXCL1, CXCL6 and SERPINE1 gene in each sample. To analyze the association of CXCL8, CXCL1, CXCL6 and SERPINE1 gene with IPM and MPLC, Panel (B) shows the results of univariate logistic regression. Panel (C) presents the mRNA expression difference of CXCL1 and CXCL8 in IPM and MPLC tumor tissues, respectively. Panels (D) provides the mean area under the curve (AUC) scores for CXCL1 and CXCL8, which can distinguish between IPM and MPLC. The predictive value of CXCL1 and CXCL8 in assessing MPLC and IPM was assessed using the logistic regression method. In Panel (E), the odds ratio of CXCL1 is presented, while Panel (F) displays the odds ratio of CXCL8 as determined through a computer verification model employing 1000 random sampling iterations. Additionally, Panel (G) illustrates the mean area under the curve (AUC) scores for CXCL1 and CXCL8 in the validated model, serving the purpose of predicting IPM and MPLC, respectively.
Discussion
In clinical practice, distinguishing between MPLC and IPM is a critical aspect of managing multiple lung lesions. The M-Mcriteria were used to classify patients as having MPLC or IPM based on histopathological examination and clinical information. Specifically, lesions were classified as MPLC if they were distinct primary tumors based on histological differences and spatial separation. IPM was diagnosed if multiple lesions were found to have the same histological type and appeared to be metastatic spread from a single primary tumor. However, the conventional diagnostic approach leads to a certain proportion of misdiagnoses and mistreatments. In response to these challenges, the International Association for the Study of Lung Cancer (IASLC) has proposed clinical and pathological standards to address the limitations of individual criteria. Since 2013, the ACCP diagnostic criteria have also incorporated molecular genetics diagnosis to aid in this differentiation. Various molecular assessment methods, such as aCGH, 15 loss of heterozygosity (LOH),16,17 DNA mate-pair sequencing (MPseq), 18 and other molecular evaluations (including p53, CEA, CK19, Ki-67, Hup-1, PE-10 19 ), have revealed molecular-level differences between IPM and MPLC. Thus, by integrating genetic profiles and M-M criteria, discrepancies in patient classification can gradually be addressed. For instance, patients initially classified as MPLC were re-evaluated using comprehensive genomic and transcriptomic data. Those with shared trunk mutations across lesions were reclassified as IPM. This genomic evidence provided additional support for resolving ambiguous cases where histopathological criteria alone were insufficient.
With the widespread use of NGS, genome research has made rapid advancements, significantly facilitating the exploration of tumor occurrence mechanisms. It also holds a pivotal role in research related to multiple nodular lung cancer. In our investigation, we observed a somatic mutation frequency of EGFR in multiple nodular lung cancer (60%), which is notably higher than the incidence reported in the Western population by TCGA.20,21 Furthermore, our study identified a low tumor mutation burden (TMB) in MPLCs, with a median TMB of 2.1. This finding aligns with the TMB of 1.92 reported by Hus et al.'s study. 22 The potential explanation for this phenomenon could be the higher representation of females and non-smoking patients in our study population. Genetic characteristics exhibit distinctions between MPLC and IPM, with IPM demonstrating a landscape of high mutation consistency.16,22–24 Some driver gene mutations, such as EGFR, BRAF, and TP53, which are considered hallmark mutations of MPLC, exhibit high mutation frequencies. 24 This concurs with Wang et al.'s study, 16 while MPLC displays a diverse range of mutations, including KRAS, MDM2, KMT2D, SDHA, and ZFHX4, which are unique to MPLC patients. Our results revealed that the genetic profiling, including the identification of shared mutations, played a crucial role in clarifying cases with intermediate characteristics by M-M criteria. Similarly, this idea was demonstrated in a recently published study by Yao et al. 25 This approach helps in refining the diagnosis and improving the accuracy of distinguishing between MPLC and IPM. Survival analyses supported the prediction efficacy of the integration classification which combining M-M criteria and mutational profiling. Furthermore, our investigation delves into the origin of lesions at the transcriptome level, enabling a comprehensive and systematic evaluation of lesion characteristics.
Chemokines and their receptors play pivotal roles in the tumor microenvironment, exerting significant influence on tumour development, progression, and metastasis. CXCL1 (also known as growth-related oncogene protein-alpha, GRO-α, or melanoma growth-stimulating activity factor, MGSA) and CXCL8 (interleukin-8, IL-8) are expressed at high concentrations within the tumour tissue of NSCLC. 26 In breast cancer, CXCL1 has been observed to recruit neutrophils, prompting them to produce and store vascular endothelial growth factor (VEGF), thereby effectively promoting tumour angiogenesis. 27 High levels of CXCL1 expression in NSCLC tumour tissue have also been independently associated with a poor prognosis. Additionally, elevated CXCL8 mRNA levels are closely linked to advanced NSCLC, distant lymph node metastasis, shorter survival times, and early recurrence.28,29 CXCL8 binds to CX-C chemokine receptor 1 (CXCR1) and CXCR2 on various cells, including neutrophils, monocytes, endothelial cells, and cancer cells, initiating cytokine and/or growth factor cascades that promote epithelial-mesenchymal transition (EMT) and thus tumour growth via autocrine feedback loops. Furthermore, the upregulation of circulating CXCL8 before clinical diagnosis has been reported as a predictor of lung cancer risk,30,31 implying that serum CXCL8 protein levels or tumour mRNA levels may serve as effective biomarkers for monitoring tumour occurrence and recurrence in lung cancer patients. Our findings establish a close relationship between the expression of CXCL1 and CXCL8, both involved in the EMT process, and the occurrence and metastasis of NSCLC tumors. This data further underscores the crucial role of chemokines in promoting tumour growth and metastasis.
Tumor-infiltrating lymphocytes (TILs) assume a pivotal role in the tumour microenvironment.32,33 The density, type, and proportion of TILs reflect the condition of the local tumour microenvironment, 34 which is linked to immune evasion in advanced cancer cells.32,33 To explore the distinctions in TILs between IPM and MPLC, we conducted an analysis of the infiltration levels of 28 immune cell subpopulations and the enrichment status of 29 immune gene sets in the tumour microenvironment using ssGSEA. Our findings reveal that IPM patients exhibit significantly higher levels of infiltrating cells, including T2 helper cells, when compared to MPLC patients. Additionally, greater granulocyte trafficking and Th1 signaling were observed in IPM patients. Natural killer cells play a role in inhibiting tumour progression and blood metastasis, 35 while cytotoxic CD4+ T cells are directly involved in tumour destruction across various cancers. External factors, such as cytokines, can promote the differentiation of cytotoxic CD4+ T cells. 36 However, research indicates that eosinophils can facilitate tumour cell migration without impacting tumour cell survival. Increased eosinophil infiltration and the secretion of CCL6 recruit tumour cells through CCL6-CCR1 signal transduction, inducing tumour cell migration and promoting eosinophil-induced tumour metastasis. Our study further investigates whether eosinophils and chemokines CXCL1/8 yield similar effects. In contrast to some reports, the genetic and immune heterogeneity differences between IPM and MPLC patients were relatively modest. This is primarily attributable to the limited number of patient cases analyzed, with only 11 cases under investigation. 37 Consequently, our study necessitates further expansion and validation.
Despite a slightly less favorable prognosis in terms of OS and DFS in both the IPM and MPLC groups, as indicated by our selected NGS-RNA grouping, there was no statistically significant difference. However, when we expanded the analysis to include pIPM, a statistical difference in OS and DFS emerged. This finding aligns with the report by Asmar MDs, 38 possibly due to the limited number of patients we included and the relatively short follow-up period. In future research, we intend to increase the sample size to further validate these observations.
Nevertheless, our study does have certain limitations. Firstly, given the complexity and rarity of distinguishing between IPM and MPLC, and the availability of suitable patient samples and strict inclusion criteria, the sample size of this study was small, so the small sample size restricts the generalizability of our findings. Most IPM patients in our study exhibited more than one clinically significant mutation for lung metastasis, possibly due to our stringent screening criteria. To address this limitation, we plan to employ whole exome sequencing and transcriptome sequencing to obtain a comprehensive overview of the mutation landscapes of the lesions, supplementing the limited number of mutations detected by NGS panel testing, which can complicate interpretation. For example, in cases where a patient shares a pair of unknown gene mutations, only MPLC can be definitively identified. Secondly, we intend to develop an algorithm that combines imaging, pathological examination, and NGS testing to enhance confidence in distinguishing between MPLC and IPM diagnoses. We believe that this approach will significantly enhance interpretation accuracy and may become routine clinical practice in the future. Thirdly, regarding the identification of biomarkers, we innovatively screened two markers (CXCL1/CXCL8); however, due to limited transcriptome data and a short patient follow-up period, we were unable to comprehensively validate their utility. Additionally, the limited number of patients included in this study and the imbalance in the number of patients with MPLC and IM may have introduced bias in the analysis. Therefore, in the future, we will expand the sample size and extend the follow-up duration to further assess their feasibility as survival and prognostic indicators.
Conclusion
NGS, based on genome and transcriptome analysis, plays a pivotal role in the identification of IPM and MPLC. In this study, we unveiled genomic distinctions between lung metastasis and multiple primary lung cancer, with the latter exhibiting a more diverse array of genetic mutations. And integration classification of M-M criteria and mutational profiling displayed prominent prediction efficacy of survival outcomes in patients. Additionally, at the transcriptome level, IPM demonstrated stronger associations with immune regulation and EMT pathways. Furthermore, CXCL1 and CXCL8, both from the EMT pathway, were identified as promising and easy to implant biomarkers distinguishing IPM from MPLC under the M-M criteria.
Supplemental Material
sj-docx-1-cbm-10.1177_18758592241308730 - Supplemental material for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers
Supplemental material, sj-docx-1-cbm-10.1177_18758592241308730 for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers by Ao Liu, Tianlin Sun, Tong Qiu, Yunqing Chen, Huiyang Qi, Wenxing Du, Zhe Wu, Zhan Huang, Wenqing Su, Changbin Zhu and Wenjie Jiao in Cancer Biomarkers
Supplemental Material
sj-xlsx-2-cbm-10.1177_18758592241308730 - Supplemental material for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers
Supplemental material, sj-xlsx-2-cbm-10.1177_18758592241308730 for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers by Ao Liu, Tianlin Sun, Tong Qiu, Yunqing Chen, Huiyang Qi, Wenxing Du, Zhe Wu, Zhan Huang, Wenqing Su, Changbin Zhu and Wenjie Jiao in Cancer Biomarkers
Supplemental Material
sj-tif-3-cbm-10.1177_18758592241308730 - Supplemental material for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers
Supplemental material, sj-tif-3-cbm-10.1177_18758592241308730 for CXCL1 and CXCL8: Reliable and feasible biomarkers differentiating intrapulmonary metastasis from multiple primary neoplasms in non-small cell lung cancers by Ao Liu, Tianlin Sun, Tong Qiu, Yunqing Chen, Huiyang Qi, Wenxing Du, Zhe Wu, Zhan Huang, Wenqing Su, Changbin Zhu and Wenjie Jiao in Cancer Biomarkers
Footnotes
Ethics approval and consent to participate
This work has been carried out in accordance with the Declaration of Helsinki (2000) of the World Medical Association. This study was approved by Ethics Committee of the Affiliated Hospital of Qingdao University. This article is a retrospective study. Therefore, the Institutional waived the requirement to obtain distinct written informed consent from the patients.
Authors’ contributions
Ao Liu and Wenjie Jiao carried out the studies, participated in collecting data, and drafted the manuscript. Wenjie Jiao, Tianlin Sun and Tong Qiu performed the statistical analysis and participated in its design. Yunqing Chen, Huiyang Qi, Wenxing Du, Zhe Wu, Zhan Huang, Wenqing Su and Zhangbin Zhu participated in acquisition, analysis, or interpretation of data and draft the manuscript. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.