
Abstract
Interstitial lung disease (ILD) has been recognized as a frequent manifestation associated with a substantial morbidity and mortality burden in patients with autoimmune rheumatic disorders. Serum autoantibodies are considered good biomarkers for identifying several subsets or specific phenotypes of ILD involvement in these patients. This review features the role of several autoantibodies as a diagnostic and prognostic biomarker linked to the presence ILD and specific ILD phenotypes in autoimmune rheumatic disorders. The case of the diverse antisynthetase antibodies in the antisynthease syndrome or the anti-melanoma differentiation-associated 5 protein (MDA5) antibodies as a marker of a severe condition such as rapidly progressive ILD in patients with clinically amyopathic dermatomyositis are some of the associations herein reported in the group of myositis spectrum disorders. Specific autoantibodies such as the well-known anti-topoisomerase I (anti-Scl70) or the anti-Th/To, anti-U11/U12 ribonucleoprotein, and anti-eukaryotic initiation factor 2B (eIF2B) antibodies seems to be specifically linked to ILD in patients with systemic sclerosis. Overlap syndromes between systemic sclerosis and myositis, also have good ILD biomarkers, which are the anti-PM/Scl and anti-Ku autoantibodies. Lastly, other not so often reported disorders as being associated with ILD but recently most recognized as is the case of rheumatoid arthritis associated ILD or entities herein included in the miscellaneous disorders section, which include anti-neutrophil cytoplasmic antibody-associated interstitial lung disease, Sjögren’s syndrome or the mixed connective tissue disease, are also discussed.
Introduction
Interstitial lung disease (ILD) is a significant disease burden in autoimmune rheumatic disorders. In fact, ILD is a substantial morbidity and mortality burden across many autoimmune rheumatic disorders. 1 Poor survival and impaired quality of life are most notable in patients with acute or chronic progressive ILD, which is often observed in patients with myositis spectrum disease, systemic sclerosis (SSc) or rheumatoid arthritis (RA). The management of ILD in patients with autoimmune rheumatic disorders is challenging due to substantial heterogeneity in disease behavior. 2 The onset and clinical course, treatment responses, and outcomes are highly variable and are often unpredictable. The treatment is not required for all patients with ILD, but it is critical to identify patients with a progressive phenotype early in the course of ILD and to introduce adequate treatment in ‘high-risk’ patients timely for improvement of outcomes. 3 The mainstay of treatment of autoimmune rheumatic disorder-associated ILD is immunomodulatory regimes, including corticosteroids, immunosuppressants, and molecular-targeted drugs such as rituximab and tocilizumab, while anti-fibrotic treatment has recently been shown to prevent progression of ILD in patients with SSc and a variety of autoimmune rheumatic disorders with progressive fibrosing ILD.4,5 There remains a critical unmet need to clarify when and in whom to initiate treatment, and which treatment regimens to choose to achieve optimal outcomes. To pursue individualized management, it is critical to predict future clinical course, treatment response, and outcomes accurately early in the course of the disease. In clinical practice, the observed disease course, patient-reported outcomes, pulmonary function testing, ILD extent and pattern on imaging, and extra-thoracic disease activity of autoimmune rheumatic disorders are used to predict subsequent disease behavior. Recently, there has been accumulating evidence for indispensable roles of serum autoantibodies in predicting risk for progression of ILD in various autoimmune rheumatic disorders, especially myositis spectrum disease. 6 This review highlights how to utilise serum autoantibodies as diagnostic and prognostic biomarkers in a patient care of autoimmune rheumatic disorder-associated ILD.
Autoantibodies as a useful diagnotic and prognostic biomarker
One of the characteristic features of autoimmune rheumatic disorders is the presence of circulating autoantibodies reactive with various cellular components, including nuclear and cytoplasmic antigens and secreted proteins. 7 Many autoantibodies are disease-specific, and are usually present at the onset and even preceding the onset of the disease. The patients do not switch from one antibody to another during the course of the disease, and autoantibodies are almost mutually exclusive. Thus, autoantibodies have been incorporated into disease classification criteria,8 –10 and are convenient biomarkers for assisting in the diagnosis in many autoimmune rheumatic disorders. The autoantibodies are also useful in disease subsetting, which offers the opportunity to predict future outcomes early in the disease course, because of their strong associations with disease manifestations, prognosis and treatment responses. There is increasing awareness of autoantibodies associated with more severe disease, which may benefit from a more aggressive treatment approach.
Current screening and diagnostic tools for ILD include clinical assessment of respiratory symptoms, auscultation, imaging studies such as chest X-ray and high-resolution computed tomography (HRCT), pulmonary function testing, and exercise-induced oxygen desaturation.3,11 A rationale for measuring convenient diagnostic serum biomarkers is to choose patients with a high risk for developing ILD before costly and/or invasive examinations. General risk factors for the development of ILD in patients with autoimmune rheumatic disorders include older age, African race, smoking and male sex.12,13 In addition, the presence of certain autoantibodies is widely used as diagnostic biomarkers, such as antisynthetase and anti-melanoma differentiation-associated 5 protein (MDA5) antibodies in myositis spectrum disorders, anti-topoisomerase (topo) I antibodies in SSc, and high-titre rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) in RA. Chest HRCT should be considered in patients with those risk factors for developing ILD in patients with autoimmune rheumatic disorders, regardless of the presence or absence of respiratory symptoms. 1
Interstitial pneumonia with autoimmune features (IPAF) is a research classification of patients with ILD who have features of autoimmunity, but are not classifiable as characterizable autoimmune rheumatic disorders, proposed by the European Respiratory Society/American Thoracic Society Task Force. 14 The classification criteria for IPAF consist of three domains: a clinical domain (extrathoracic features), a serological domain (circulating autoantibodies), and a morphological domain (chest imaging, histopathological, or pulmonary physiological features), and at least one feature from at least two of the three domains is required for the classification. A serological domain includes anti-nuclear antibody (ANA) ⩾1:320 with diffuse, speckled, and/or homogeneous patterns or any titer in case of nucleolar or centromere pattern; RF ⩾2 × upper limit of normal; and disease-specific autoantibodies such as antisynthetase, anti-MDA5, anti-topo I antibodies and ACPA.
The clinical course of ILD in autoimmune rheumatic disorders is highly variable. Some patients show rapid or slow progression leading to respiratory insufficiency, but others have a stable course with no clinically meaningful progression. Even in patients with stable ILD, some develop acute exacerbations. Specifically, organizing pneumonia often found mainly in patients with RA resolves completely by short-course treatment with corticosteroids or even spontaneously. Acute or subacute ILD in patients with dermatomyositis is sometimes treatment-resistant and fatal, especially in those with anti-MDA5 antibody, but those in patients with antisynthetase antibody usually respond to immunosuppressive treatment, but recur frequently. In contrast, a worse prognosis is observed in patients with chronic ILD with progressive, irreversible fibrotic disease, resembling the clinical course of idiopathic pulmonary fibrosis (IPF). 15 This phenotype is now called progressive fibrosing ILD, which is characterized by progressive pulmonary fibrosis associated with worsening respiratory symptoms, decline in pulmonary function, decreased quality of life, and risk of early death despite treatment. 16 Proportions of patients with PF-ILD are variable among underlying autoimmune rheumatic disorders, and range from 16% to 40%. 17 Two predominant autoimmune rheumatic disorders with progressive fibrosing ILD are RA and SSc. 3 In clinical practice, detection of PF-ILD early in the course of the disease is critical, but it is often difficult. In such cases, longitudinal observation is recommended, and the definition of progressive fibrosing ILD has been proposed as a relative decline of forced vital capacity (FVC) % predicted ⩾10% alone or relative decline of FVC % predicted 5–10% in combination with at least one of relative decline of diffusing capacity of the lung for carbon monoxide (DLCO) % predicted ⩾15%, increased extent of fibrosis on HRCT, or worsening respiratory symptoms within 24 months despite standard treatment of ILD. 18 Nevertheless, it is better to predict the clinical course of ILD early in the disease course without impaired pulmonary function. In this regard, identification of prognostic biomarkers for progressive fibrosing ILD should contribute to early detection of progressive cases and guidance of treatment decision. Therefore, clinical and biological factors that predict subsequent progression of ILD have been extensively investigated in individual autoimmune rheumatic disorders. For this purpose, serum autoantibodies are widely used as a prognostic biomarker.
Key autoantibodies associated with ILD in autoimmune rheumatic disorders are summarised in Table 1. Autoantibodies are routinely used in patients’ care of ILD in patients with autoimmune rheumatic disorders as a diagnostic biomarker to pursue extensive ILD screening as well as a prognostic biomarker to predict future progression and prognosis and guide the management plan.
Key autoantibodies associated with interstitial lung disease.
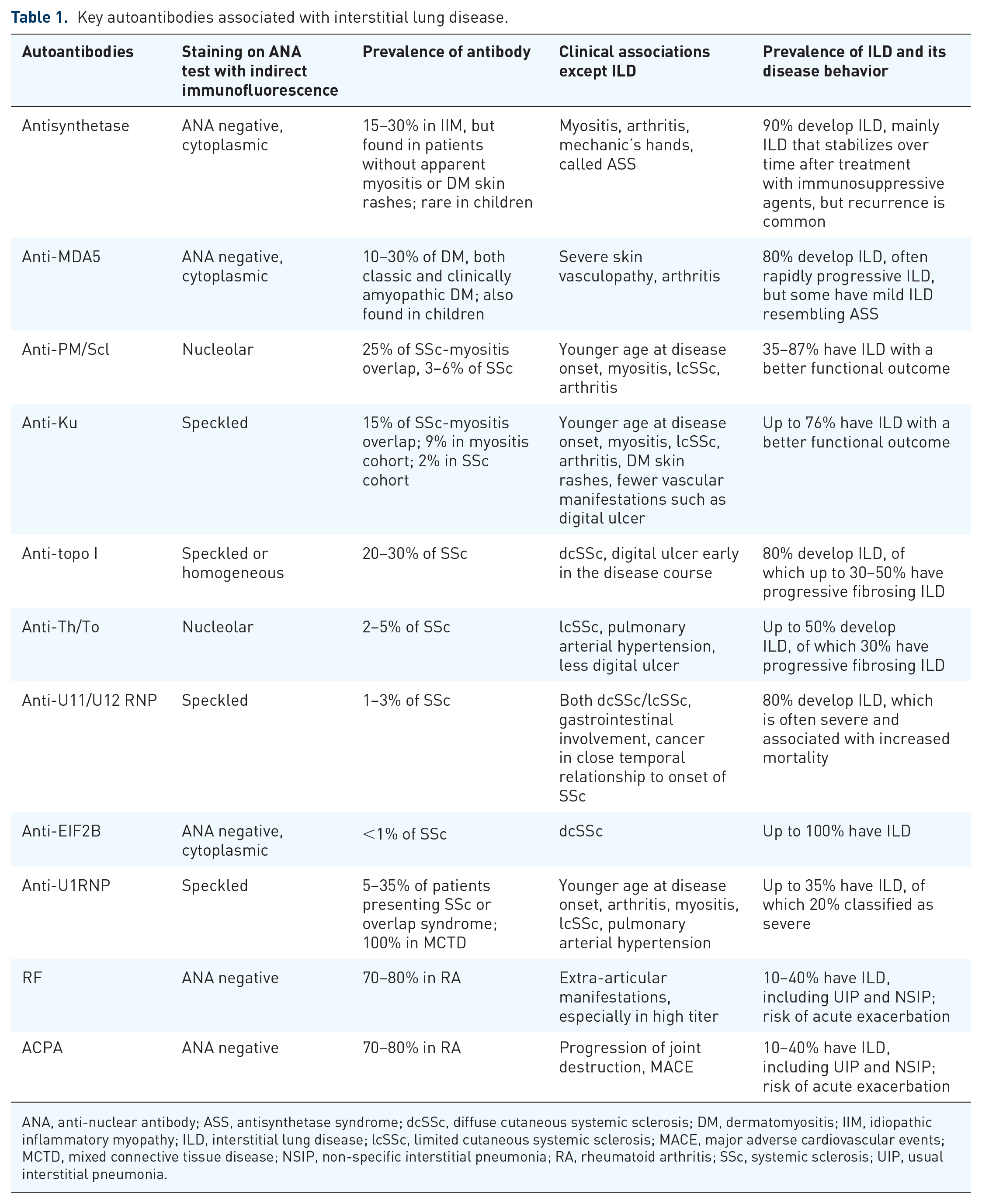
ANA, anti-nuclear antibody; ASS, antisynthetase syndrome; dcSSc, diffuse cutaneous systemic sclerosis; DM, dermatomyositis; IIM, idiopathic inflammatory myopathy; ILD, interstitial lung disease; lcSSc, limited cutaneous systemic sclerosis; MACE, major adverse cardiovascular events; MCTD, mixed connective tissue disease; NSIP, non-specific interstitial pneumonia; RA, rheumatoid arthritis; SSc, systemic sclerosis; UIP, usual interstitial pneumonia.
Myositis spectrum disorders
The group of inflammatory conditions known as myositis are heterogeneous disorders with an autoimmune basis in which characteristic muscle inflammation is usually present. These disorders are divided into five main types: dermatomyositis, polymyositis, overlap myositis, sporadic inclusion body myositis, and immune-mediated necrotising myopathy. 19 Multiple phenotypes are included within the broad term ‘myositis’ and paradoxically, some of them do not include muscle involvement, although from the clinical and immunological viewpoints they are grouped in the same box. That is why they are often referred to as ‘myositis spectrum disorders’.
Several antibodies directed against autoantigens have been identified in myositis patients. These are usually classified into specific (only appearing in patients with myositis) or associated (can appear in patients with myositis and in other similar conditions). Recently, the term ‘myositis-spectrum disease antibodies’ was coined broadly to define these autoantibodies. 20 They are more biomarkers of certain specific features or clinical manifestations of these conditions, such as lung involvement, than markers of myositis itself. For example, as will be seen later, some patients positive for anti-Jo1 antibody, a typical myositis-specific autoantibody, may present with ILD alone, without myositis.
The lung is one of the organs most often affected in patients with myositis spectrum disorders, with or without muscle involvement, 21 reaching a prevalence of 41% in a recent analysis. 22 Furthermore, lung involvement is the leading cause of mortality in this group of systemic diseases, although it has a highly variable clinical course and prognosis, consistent with the heterogeneous characteristics of myositis patients. The presence of several autoantibodies may be of value to establish an accurate diagnosis and estimate the prognosis of this feature.
The following sections will attempt to provide a comprehensive description of the various antibody-associated myositis phenotypes, with particular focus on the diagnosis and prognosis of these conditions.
Antisynthetase syndrome (ASS)
In daily clinical practice, clinicians may encounter a patient with several clinical manifestations, such as arthritis, hiker feet [Figure 1(a)] or mechanic’s hands [Figure 1(b)], shortness of breath and perhaps, muscle weakness (e.g. when attempting to rise from a chair or combing hair). The astute physician, after noting bibasal inspiratory crackles on lung auscultation, could suspect the diagnosis of ILD in a patient with inflammatory muscle disease. Pulmonary function tests showing a reduction in the FVC and DLCO, HRCT scans with features of ILD (e.g. ground glass opacities, lung fibrosis, reticular pattern), and if necessary, muscle biopsy findings will confirm the suspected diagnosis. However, it is the autoantibody profile that will enable better classification of the patient’s clinical condition. In this case, positive testing for anti-histidyl-tRNA synthetase (referred to as anti-Jo1 antibody) the most common antisynthetase antibody described to date, will provide relevant information regarding the patient’s prognosis and outcome.

Hiker feet (a: arrow) and mechanic’s hands (b: arrow) observed in a patient with antisynthetase syndrome.
Eight different autoantibodies against tRNA synthetase have been described (Table 2) and they are associated with specific clinical features and with different outcomes regarding survival or mortality.23 –25 Analysis of large multicenter cohorts of patients with anti-Jo1 antibodies has enabled depiction of the long-term clinical outcome. In a retrospective longitudinal study including 148 patients with anti-Jo1 ASS and a follow-up of 78.3 months, the authors found that the disease mainly acted as a chronic condition in terms of lung involvement. 26 Only a small percentage of patients reached the composite endpoint defined ad-hoc for the study: death due to respiratory failure directly related to ASS, long-term oxygen therapy requirement, or lung transplantation.
Antisynthetase antibodies.

When analysing the less common antisynthetase antibodies, we should be aware of the potential for selection bias. As ASS is uncommon, the small available series may favour the bias of assigning some clinical features to specific autoantibodies. This was the case of anti-threonyl-tRNA synthetase, known as anti-PL7 (PL stands for precipitation line where the antibody was first identified in the laboratory). 27 In a study including a small case series, anti-PL7 seemed to be a marker of pericardial disease, but the association disappeared when large patient cohorts were analysed.
A recent study including the largest cohort of patients with ASS analysed to date has shed light on this topic. 28 In total, 828 ASS patients from 10 countries and 63 hospitals were included in the study, which provided relevant data regarding the rarer antisynthetase antibodies, such as anti-EJ (38 patients included) and anti-OJ (18 patients included). Comparisons between the different groups with antisynthetase antibodies found that arthritis was more frequent in those with anti-Jo1 than in those without. Moreover, in some antibody specificities, such positive status to anti-PL7 or anti-PL12, the diagnostic delay was greater than in the anti-Jo1 antibodies group, likely because certain atypical forms of the syndrome had been misdiagnosed. The acute form, which is unusual in patients with ASS, was more frequent in patients positive for anti-EJ antibodies. In addition, the authors stated that the clinical presentation was quite similar between the various antibody specificities at the end of follow-up (i.e. it did not differ from one specificity to another). Finally, and most importantly, survival was not influenced by the underlying antisynthetase antibody (at least after 5 years of follow-up), which agrees with findings from other studies.29,30 The cumulative 10-year survival rate of the ASS patients is estimated at nearly 75%.24,26
Although most patients with ASS will develop a chronic form of ILD that stabilizes over time after treatment with immunosuppressive agents, some factors associated with the syndrome have been recognized as indicators of a poor prognosis. Older age, and several serological factors, such as increased ferritin concentration, coexistence of other autoantibodies (e.g. anti-Ro52 or anti-Ro60), or a phenotype without muscle involvement, seem to be related to a poorer lung function outcome. Coexistence of antisynthetase autoantibodies (mainly anti-Jo1) and anti-Ro52 antibodies has been identified as marker of more severe ILD and a poorer prognosis for lung function outcome in adults and in juvenile forms. Researchers in France analysed a cohort of 89 consecutive anti-Jo1 patients with ASS and found that those who also tested positive for anti-Ro52 more commonly exhibited symptomatic ILD and higher fibrosis scores on HRCT. 31 Similar results were found by Váncsa et al. 32 Other researchers have reported that the presence of the Ro52-Jo1 complex is a biomarker of a more acute form of ILD and of refractoriness to conventional immunosuppressive therapy, but not to rituximab, 33 although these data have not been replicated by other groups. Lastly, in an analysis of a large cohort of 302 patients with juvenile dermatomyositis, Sabbagh et al. 34 found that the coexistence of Ro52 with anti-Jo1 was associated with more severe disease and a poorer prognosis.
The anti-PM/Scl phenotype
The anti-PM/Scl autoantibody is directed against a multi-subunit nucleolar protein complex known as exosomes. These are endocytic vesicles containing RNA and proteins that are involved in RNA degradation and processing. Although the PM/Scl antigen is a particle with 11 different polypeptides, it is believed that the autoantibodies recognize the 75-kD or preferentially, the 100-kD protein. Thus, most commercial tests for anti- PM/Scl antibodies contain the 100-kD antigen.
The clinical phenotype of patients with anti-PM/Scl may resemble that of patients with ASS, probably because of a shared genetic background.35,36 At times manifestations of myositis, arthritis, and even scleroderma features such as Raynaud’s phenomenon and specific capillaroscopy findings may make it difficult to differentiate between ASS and the anti-PM/Scl scleromyositis phenotype until the immunological profile results are obtained. Lung involvement in anti-PM/Scl-positive patients is common, documented in 35-87% of patients in the reported series.37,38 But the main point to keep in mind is that ILD seems to have a better functional outcome in these patients. Two studies have addressed this topic. The first, carried out by researchers in Barcelona, 39 analysed progression-free survival in patients with systemic sclerosis and ILD according to positive status to anti-PM/Scl antibodies. Progression-free survival in the study was defined as the time period in which ILD was stable up to the point when an FVC decrease of at least 10% from baseline or death occurred over follow-up. When patients positive for anti-PM/Scl antibody were compared with SSc patients negative for anti-PM/Scl but positive for anti-topo I, progression-free survival at 10 years’ follow-up was higher in anti-PM/Scl-positive than anti-topo I-positive patients (76% versus 29%, respectively). Other studies focused on the clinical features of patients with anti-PM/Scl antibodies have reported similar results,40 –42 thus defining a characteristic phenotype in which ILD is present and has a good outcome. The second study, a recent multicenter report from the EUSTAR database, complemented with a case–control study from another center, found that lung function outcome was better in ILD patients positive for anti-PM/Scl. 43
Anti-Ku syndrome
Patients with anti-Ku syndrome may also develop a type of overlap condition between SSc and myositis. A recent cluster analysis in a cohort of 42 consecutive patients positive for anti-Ku antibodies disclosed a high prevalence of ILD when associated with myositis (high creatine kinase levels). 44 Data from a large cohort of Chinese patients with myositis revealed that only 1.73% were anti-Ku positive, but 76.2% of the anti-Ku-positive patients developed ILD. 45 As occurs in anti-PM/Scl-positive patients, it seems that the ILD that accompanies anti-Ku antibodies is not severe; nonetheless, the short reported series preclude strong statements.
Anti-MDA5-positive, dermatomyositis-associated, rapidly progressive ILD
In 2005 Sato et al. 46 described a new autoantibody in patients with dermatomyositis. The autoantibody was found to be a good biomarker of severe disease; that is, the rapidly progressive ILD that can develop in patients with the amyopathic or hypomyopathic form of dermatomyositis. The target antigen recognized by this autoantibody was later identified as MDA5, also known as interferon-induced helicase C domain-containing protein 1 (IFIH1), which participates in a well-recognized innate antiviral response, triggering the production of type I interferons on detection of viral double-stranded RNA at the cytosol. MDA5 is a member of the retinoic-acid inducible gene (RIG)-I-like helicases, which act as cytoplasmic RNA sensors. 47
The clinical relevance of anti-MDA5 antibodies resides in their capacity to identify patients with dermatomyositis – to date the only myositis subtype related with the antibody – who may have rapidly progressive ILD and a poor prognosis. Up to 80% of these patients do not survive even after a prompt diagnosis and intensive immunosuppressive therapy. 48 The anti-MDA5 antibody currently detected by commercial tests enables the identification dermatomyositis patients at risk of rapid lung function impairment: worsening of radiological interstitial changes with progressive dyspnoea and hypoxemia within 1 month after the onset of respiratory symptoms. 49 Imaging detection of reticular opacities, ground-glass opacities, or a honeycomb pattern may assist the diagnosis. Several studies worldwide have confirmed the role of these autoantibodies as a biomarker of this severe condition.50 –52 A meta-analysis carried out by researchers from China concluded that detection of anti-MDA5 antibodies was a valuable tool to identify dermatomyositis patients (mainly those with the amyopathic form) who will develop rapidly progressive ILD with a poor prognosis. 52
The next question is whether all anti-MDA5-positive dermatomyositis patients will develop this severe condition, and the answer is no. Some researchers have found that the clinical spectrum in patients with anti-MDA5 antibodies may expand to other features or that some patients may develop a milder form of ILD similar to that seen in the anti-PM/Scl phenotype or ASS,50,51 discussed above. Moreover, arthritis and skin lesions can be a hallmark of other phenotypes of the disease. Researchers from the French Myositis Network recently reported a study 53 characterizing several phenotypes related to anti-MDA5. The main phenotype (only 18% of the total cases reported) corresponded to the severe condition of rapidly progressive ILD, but two additional phenotypes were defined, one in which patients only have skin lesions and arthritis/arthralgia with a good prognosis (55%), and the other characterized by severe skin vasculopathy and myositis, with an intermediate prognosis.
At this point we must face the challenge of how to manage a dermatomyositis patient testing positive for anti-MDA5 antibodies. We first need to know whether or not the patient will develop rapidly progressive ILD. Some reported data may help in addressing this difficult topic. The anti-MDA5 antibody level,54,55 ferritin concentration, 56 and presence of anti-Ro52 antibodies57,58 should all be considered. Focusing on the coexistence of anti-Ro52 antibodies, a well-known situation in patients with ASS, the available data indicate a role of this biomarker for determining the prognosis in patients with anti-MDA5 antibodies. Ro52 is an interferon-inducible protein of the tripartite motif family (TRIM21), usually located in the cytoplasm, that regulates inflammation induced by the interferon pathway. 59 The association of anti-Ro52 with anti-MDA5, which is not a rare occurrence, seems to identify a more aggressive phenotype (i.e. rapidly progressive ILD), as has been reported in a study investigating 83 patients with anti-MDA5-positive, clinically amyopathic ILD. 57 Coexistence of anti-Ro52 in these patients was significantly associated with a high percentage of rapidly progressive ILD cases. The same results were observed in a retrospective analysis of a cohort of Japanese patients with myositis: those with anti-MDA5 plus anti-Ro52 antibodies had a poor prognosis. 58 Although the association of anti-Ro52 with anti-MDA5 and with ASS has been described in juvenile forms of dermatomyositis, data on its contribution as a marker of a poor prognosis are not available in this population. 34
Anti-SAE antibodies
In 2007, Betteridge et al. 60 described a new autoantibody in a few dermatomyositis patients. It was directed against a 40-kD and 90-kD doublet of bands on protein immunoprecipitation and was later identified as small ubiquitin-like modifier 1 activating enzyme A (SAE), a protein involved in post-translational changes. Two of 20 dermatomyositis patients who were positive for anti-SAE antibody had associated non-specific ILD. In two reported case series,61,62 57–71% of anti-SAE-positive dermatomyositis patients had ILD. In our hospital dermatomyositis cohort, 5% of patients are anti-SAE positive and all have some degree of ILD. Although this factor has not been studied in large cohorts, it seems that anti-SAE could be a marker of a dermatomyositis phenotype in which ILD is not rare and does not have a poor outcome.
Systemic sclerosis (SSc)
SSc is a systemic autoimmune disease characterized by microvasculopathy, immune dysregulation, and excessive fibrosis, but detailed pathogenesis still remains unclear. 63 In addition to characteristic skin and peripheral vascular involvement, SSc can affect multiple organ systems, including the lung, heart, kidney, and gastrointestinal tract. ILD is one of the most important causes of mortality in SSc patients along with pulmonary arterial hypertension. 64 Disease behavior of SSc-ILD is highly various, and only <30% of the patients have progressive fibrosing ILD, leading to respiratory insufficiency.65 –67 Most patients who develop severe restrictive lung disease experience ILD progression prominently in the first 5 years following the onset of SSc. The most common initial symptoms of SSc-ILD are exertional dyspnea and dry cough, although early SSc-ILD can be asymptomatic. Therefore, recent evidence-based European consensus statements for SSc-ILD recommend chest HRCT for screening of ILD at diagnosis of SSc. 11 A HRCT pattern suggestive of fibrotic non-specific interstitial pneumonia (NSIP), which shows bilateral ground-glass opacities, reticulation, and traction bronchiectasis most prominent in the lower lobes, is typical of SSc-ILD, while cellular NSIP or usual interstitial pneumonia (UIP) patterns are observed in a small number of cases. 12 Despite the apparent association of ILD and morbidity and mortality in patients with SSc, screening for ILD and monitoring for disease progression is still challenging in the clinical setting.
Autoantibodies in SSc
Anti-nuclear antibodies detected by the indirect immunofluorescence technique are a hallmark of SSc and are found in >95% of the patients. At least 12 autoantibody specificities associated with SSc have been reported and well characterized, and >80% of SSc patients have one of these autoantibodies. 68 These include anti-topo I or anti-Scl-70; anticentromere antibody (ACA); anti-RNA polymerase (RNAP) III; anti-U3 ribonucleoprotein (RNP); anti-Th/To; anti-U11/U12 RNP; anti-centriole; anti-eukaryotic initiation factor 2B (eIF2B); anti-U1 RNP; anti-Ku, anti-PM/Scl; anti-RuvBL1/2 antibodies. SSc-related autoantibodies target various nuclear or cytoplasmic components involved in essential cellular processes, such as cell division and transcription. These autoantibodies are rarely seen in patients with other connective tissue diseases without SSc features and thus are important diagnostic markers. The detection of SSc-related autoantibodies is clinically useful in classifying SSc patients into subtypes that are almost exclusively associated with characteristic clinical phenotypes. The main autoantibodies associated with ILD include anti-topo I, anti-Th/To, anti-U11/U12 RNP and anti-eIF2B antibodies, while antibodies associated with SSc overlap syndrome, mainly with myositis, occasionally have ILD. Anti-PM/Scl and anti-Ku syndromes are covered in a section of myositis spectrum disorders.
Anti-topo I antibodies
Anti-topo I antibody is detected in 20–30% of SSc patients in many ethnic groups. Approximately two-thirds of anti-topo I-positive patients have diffuse cutaneous systemic sclerosis (dcSSc), but progression of skin thickening is slower than in those with anti-RNAP III antibody. 69 Anti-topo I antibody is associated with a high risk of severe ILD, cardiomyopathy and peripheral vascular complications, such as digital ulcer and gangrene, particularly early in the disease course.70,71 Anti-topo I antibody is considered to be a marker for poor prognosis, and patients with severe ILD die of this complication at an average of 10 years after the onset of SSc. The prevalence of ILD is comparable between dcSSc and limited cutaneous systemic sclerosis (lcSSc) patients with anti-topo I, but survivals are worse in dcSSc than in lcSSc patients.71,72 In SSc patients with anti-topo I, the concomitant presence of IgM isotype of anti-topo I antibodies is associated with subsequent disease progression. 73
Anti-Th/To antibodies
Anti-Th/To antibody occurs in patients with lcSSc, although its frequency overall in SSc patients is only 2–5%. 74 Like ACA-positive patients, anti-Th/To-positive patients are predominantly Caucasians, but tend to have a shorter duration of Raynaud’s phenomenon before the onset of other symptoms such as puffy fingers. Digital ulcer and digital gangrene are infrequent, but patients with anti-Th/To antibody can have significant ILD or pulmonary arterial hypertension, the latter often independent of ILD, which often occurs early in the disease course. This increased frequency and severity of pulmonary complications result in a decreased survival compared with lcSSc patients without this antibody.
Anti-U11/U12 RNP antibodies
Anti-U11/U12 RNP antibody is a rare antibody specificity found in 1–3% of patients with SSc. 75 This antibody recognizes RNA binding region containing 3 (RNPC-3) associated with U11/U12 RNA, and produces speckled nuclear staining on indirect immunofluorescence. Patients with this antibody are classified as having either dcSSc or lcSSc. A characteristic feature of patients with this antibody is a high frequency of ILD (>80%), which is often severe and progressive, and associated with a 2.25-fold greater risk of death in comparison with anti-U11/U12 RNP-negative patients with ILD. 75 A relationship between anti-U11/U12 RNP antibody and severe ILD has been replicated in an independent cohort, which has additionally found the association of this antibody with moderate to severe gastrointestinal involvement. 76 Recently, anti-U11/U12 RNP antibodies were found in patients with cancer in close temporal relationship to the onset of SSc. 77
Anti-eIF2B antibodies
Anti-eIF2B antibody is a novel anti-cytoplasmic antibody identified in SSc sera negative for all known SSc-related autoantibodies. 78 The prevalence was ~1% in the United Kingdom cohort, 78 but 7% in the North American cohort. 79 The majority of patients with anti-eIF2B antibodies are classified as having dcSSc and have ILD.
Screening and diagnosis of SSc-ILD
The prevalence of ILD in SSc patients varies depending on detection methods or tools; the prevalence using HRCT is estimated at 47–84%, 12 while the prevalence is <30% using auscultation, chest X-ray, or pulmonary function testing (FVC % predicted).80,81 A number of demographic and clinical characteristics are associated with a higher risk of developing ILD in SSc patients. Patients with SSc are routinely divided into lcSSc and dcSSc based on the extent of skin thickening, and ILD was more common in dcSSc than in lcSSc subset. 82 African American race and male sex were also reported to be associated with ILD. 82 Risk factors for the development of ILD also include anti-topo I antibody. Multivariate analyses suggest that the risk of developing ILD is more closely associated with anti-topo I antibody rather than the dcSSc/lcSSc subset. 82 Anti-topo I-positive patients have the highest incidence of ILD (>80%) independent of dcSSc/lcSSc subsets.70,83 A novel autoantibody targeting U11/U12 RNP is associated with severe ILD and decreased survival. 75 Other antibodies associated with ILD include anti-Th/To in the case of a nucleolar pattern on routine ANA assay 74 and anti-eIF2B in the case of a cytoplasmic pattern (a negative ANA). 78 In the context of lcSSc with severe ILD, anti-U11/U12 RNP and anti-Th/To are the most common autoantibodies. In contrast, the presence of ACA, anti-RNA polymerase III, and anti-U3RNP were associated with a low risk of ILD. 71
Prediction of progression of SSc-ILD
Once ILD has been diagnosed in patients with SSc, it is important to identify which patients are likely to progress, because some patients experience progressive loss of pulmonary function, and others progress slowly or exhibit stable disease. The prediction of subsequent progression of SSc-ILD should be considered as a first step in the management of the patients. However, at present, there are no reliable prediction models for progression of SSc-ILD. In clinical practice, the severity of SSc-ILD is staged based on the extent of ILD on HRCT and FVC. 84 A number of other clinical factors associated with the progression of SSc-ILD have been reported and include older age, smoking history, early dcSSc, progression of skin thickening, history or presence of arthritis, desaturation after 6-minute walk test, and decreased DLCO.12,85 –87 Serum biomarkers have also been extensively investigated, and elevated levels of C-reactive protein, Krebs von den Lungen (KL)-6, chemokine ligand 18 are shown to be associated with a subsequent decline of FVC and/or development of end-stage of ILD or death.66,88 –90 Several studies have reported that anti-topo I antibody may be associated with the faster progression of SSc-ILD in the short term. 91 However, its utility as a prognostic biomarker to predict the future progression of ILD still remains controversial as many observational studies did not support this finding.
Rheumatoid arthritis (RA)
RA is a systemic autoimmune disease primarily affecting synovial joints, but also involves other organ systems including the lung. 92 The lung involvement is the most common extra-articular manifestation, and includes a variety of respiratory complications, such as ILD, airway disease, pleuritis, nodulosis and vasculitis. 93 Infections and drug hypersensitivity are important differential diagnoses and should be considered in the differential diagnosis of RA patients presenting with respiratory symptoms. ILD is one of the major causes of morbidity and mortality, 94 and is associated with a significantly shortened survival in patients with RA, compared to those patients without ILD. 95 In addition, the presence of ILD often interferes with outcomes of a treat-to-target strategy for synovitis by influencing choices of disease-modifying anti-rheumatic drugs. The prevalence of ILD in RA patients varies from 10% to 40%, depending on the definition of ILD, 95 but it is a major proportion of autoimmune rheumatic disorder-associated ILD in clinical practice because RA is the most common autoimmune rheumatic disorder. Another feature of RA-ILD is high variability in HRCT patterns and lung histopathology. The most common HRCT patterns in RA-ILD are UIP and NSIP, while organizing pneumonia (OP) is less common. 96 Disease behavior is also highly heterogeneous, and some patients show progressive fibrosing ILD, but others have a stable course. Even in patients with stable ILD, some develop acute exacerbations, which is more common than other ILD associated with autoimmune rheumatic disorders. 97
RA is characterized by the presence of RF, which is reactive with the Fc portion of IgG, and antibodies reactive to autoantigens undergoing post-translational modifications, mainly by citrullination (in this case, a term ACPA is used) but also other modifications. 98 These antibodies are included in the classification of RA because of adequate disease sensitivity and specificity, whereas ACPA has greater specificity than RF with similar sensitivity. 8 These autoantibodies are thought to be pathogenic, because they might directly induce stimulatory signals by ligating key citrullinated cell surface molecules or, alternatively, act as immune complexes on Fc receptors. 98 A number of studies have revealed pathogenic processes to the onset of synovitis in RA, and production of RA autoantibodies precedes the onset of RA. 99 Specifically, in a genetically predisposed host, environmental insults, epigenetic modifications and post-translational modifications can lead to loss of immune tolerance with subsequent production of autoantibodies, including RF and ACPA. This ultimately leads to clinically overt arthritis. Therefore, the onset of arthritis is thought to be the last step of a series of pathogenic events lasting years. Mucosal surfaces, such as oral and airway mucosa, are major sites for initiating this autoantibody response. 100 Several risk factors, such as smoking, dust inhalation, bacterial colonization, and preceding lung diseases such as ILD are known as environmental triggers. Interestingly, ILD may precede the development of articular manifestations, and 14% of patients with RA-ILD had been diagnosed with ILD 1 to 5 years before the RA diagnosis. 94 In this regard, the association with MUC5B promoter variant, which was observed in patients with IPF, was also found in patients with RA-ILD and more specifically in patients with the UIP pattern on HRCT. 101
There are a number of risk factors for the development of ILD in RA patients, including older age, male sex, and history of smoking, and high-titre RF and ACPA are also recognized as diagnostic biomarkers for ILD. 102 The risk of developing ILD increases with prolonged duration of RA, especially sustained disease activity. 103 RF and ACPA, especially high levels and the IgA isotype, are associated with the occurrence of ILD in RA patients, but it is difficult to assess if they are independent risks due to many confounding factors, such as smoking, age, and sex.104 –106 Interestingly, up to 15% of patients with idiopathic ILD without synovitis have ACPA. Among them, serum ACPA positivity is predictive of developing RA in the following 5 years, suggesting common genetic and environmental predisposing factors between RA and ILD, especially IPF.107,108 Alternatively, ILD contributes to the onset of RA as one of the triggers for production of RA autoantibodies.
In terms of risk factors for progression of RA-ILD, the UIP pattern is associated with higher mortality rates, similar to IPF. 109 The prediction model combining the extent of ILD on HRCT and FVC, which was described in patients with SSc-ILD, was also useful to predict mortality in patients with RA-ILD. 110 RF is also listed as a prognostic biomarker. In this integrated analysis of 2331 patients with early RA, RF was associated with all-cause mortality as well as ILD-related death, independent of other autoantibodies, including ACPA. 111
Miscellaneous diseases
This section describes other systemic autoimmune diseases that can progress with some degree of ILD, in which associated autoantibodies may be of help to identify the phenotype.
Systemic lupus erythematosus
ILD is uncommon 112 in patients with systemic lupus erythematosus (SLE) to such an extent that if a patient diagnosed with SLE develops ILD, an alternative diagnosis should be sought (e.g. mixed connective tissue disease or Sjögren’s syndrome). In this line, certain autoantibodies, such as anti-Ro/SSA, anti-La/SSB, and anti-U1 RNP may test positive in the rare SLE patients with ILD, whereas the most specific SLE antibodies, anti-DNA antibodies, are not associated with ILD. 113 One possible exception is lupus pneumonitis. This rare manifestation (<1% of lupus patients), which usually appears in the context of a disease flare, may progress to chronic ILD due to long-term lung damage.
Sjögren’s syndrome
The prevalence of ILD in Sjögren’s syndrome is nearly 20%, with non-specific interstitial pneumonia being the most common pattern. 114 The role of anti-Ro antibodies is not well understood in these patients. Whereas anti-Ro52 antibodies are a recognized risk factor for ILD and screening by HRCT in these patients is recommended by some authors, 115 others have reported no association between anti-Ro60/Ro52 positive status and ILD. 116 Nonetheless in this latter study, a significant percentage of anti-Ro52-positive patients developed a non-usual interstitial pneumonia pattern.
ANCA vasculitis
The association between anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis and ILD has been recognized since the 1990s, specifically in older patients.117,118 Two phenotypes have been identified. The first comprises patients with ANCA-positive vasculitis, mainly microscopic polyangiitis with p-ANCA-positive antibodies (myeloperoxidase (MPO)-ANCA), that develop ILD. 119 The second includes ANCA-positive patients with no clinical features of vasculitis, but with ILD. 120 Patients within the second phenotype may develop full-blown ANCA-associated vasculitis on follow-up, but this is not always the rule. The outcome appears to depend on the fibrosis pattern, with a poorer outcome in those with UIP than in those with non-specific interstitial pneumonia. 121 ANCA testing should be included in the diagnostic work-up of ILD.
Mixed connective tissue disease
Mixed connective tissue disease (MCTD) was described in 1972 by Sharp et al. 122 ILD is a feature in up to 48% of MCTD patients.123,124 Several autoantibodies have been related with the ILD in MCTD, such as anti-Sm 125 and anti-U1 RNP antibodies,124,126 both of which are included in the spliceosome, a cytoplasmic macromolecule responsible for processing pre-messenger RNA by removing intronic sequences to obtain mature RNA that can be translated into proteins. This is consistent with the higher prevalence of ILD found in MCTD with epitope spreading during follow-up; that is, recognition of a new epitope (e.g. Sm) which is spatially related with another epitope for which the patient had autoantibodies previously (e.g. U1 RNP). 127 Anti-Ro52 antibodies are also reported to be a biomarker of ILD in MCTD patients.124,128 All three autoantibodies are considered risk factors for developing ILD in patients diagnosed with MCTD.
Conclusions
Autoantibodies are a characteristic feature of the autoimmune rheumatic disorders, and are convenient, useful diagnostic and prognostic biomarkers in clinical practice of ILD associated with autoimmune rheumatic disorders. Measurement of relevant autoantibodies contributes to precision medicine because of their utility in the prediction of future progression and prognosis of ILD and potential guidance of treatment decision. Although the measurement of ANA, RF, ACPA, and ANCA in patients with ILD lacking overt features of autoimmune rheumatic disorders is generally recommended, we suggest a more comprehensive testing for antibodies including the myositis spectrum disease autoantibodies and SSc-related autoantibodies because a good correlation with cytoplasmic staining is not always observed. However, as autoantibodies are just surrogate biomarkers, a clinical decision should be made based on comprehensive assessments, including physical findings, pulmonary function and imaging studies. In addition, there are a number of methods for the measurement of autoantibodies each with their own inherent limitations. Physicians should understand them best to interpret results in the clinical context.