
Abstract
Abscopal immunity—the regression of distant, non-irradiated lesions after localized radiotherapy (RT)—signals conversion of focal DNA damage into systemic antitumor immunity. This review advances a unifying three-stage framework—initiation, amplification, and reinforcement—explaining how RT can be leveraged to elicit durable systemic control. In initiation, immunogenic cell death and cytosolic DNA activate cGAS–STING (with TLR3–interferon (IFN)-I as a compensatory axis), driving dendritic cell recruitment and cross-priming in tumor-draining lymph nodes. Amplification entails chemokine-guided trafficking and expansion of CXCR3+ cytotoxic T cells, together with stromal and vascular remodeling that enable infiltration at out-of-field sites. Reinforcement reflects the balance between memory formation and adaptive resistance (PD-L1 upregulation, myeloid/Treg accrual, adenosine, and metabolic checkpoints), defining actionable targets for combinatorial intervention. We critically appraise clinical data showing that RT paired with immune-checkpoint inhibition can increase out-of-field control in selected settings, whereas heterogeneous or negative trials underscore the importance of dose and fractionation, field design/target coverage, RT-immune checkpoint inhibitor sequencing, and sparing of lymphoid structures. We outline emerging levers—including spatially fractionated RT, FLASH RT, proton therapy, myeloid- and adenosine-axis blockade, and nanotechnology-enabled in situ vaccination—and candidate biomarkers (interferon-response signatures, circulating tumor DNA kinetics, T-cell clonotypes). Operationalizing these principles points toward making the abscopal effect a predictable, clinically actionable endpoint rather than a rarity.
Plain language summary
Radiotherapy is designed to treat a tumor in one spot. Yet, in a small but important number of patients, tumors far from the radiation field also shrink. This rare whole-body response is called the “abscopal effect.” It occurs when radiation breaks cancer cells apart and releases “danger” signals and pieces of tumor. Nearby immune cells pick up these clues, learn what the cancer looks like, and train killer T cells that can travel through the body to attack tumors elsewhere. Why is this effect uncommon? Cancer can hide behind “brakes” on the immune system, and large radiation fields may harm helpful immune cells in nearby lymph nodes. The dose, schedule, and timing of radiotherapy also matter: the immune system needs enough time to learn, expand, and move. Doctors are testing ways to make abscopal responses more reliable. One approach combines radiotherapy with immunotherapy drugs that lift immune “brakes” (such as PD-1, PD-L1, or CTLA-4 inhibitors). Other tactics include treating a limited number of tumor sites, using precise beams to spare lymphoid tissue, and choosing doses and timing that best support immune learning. Emerging strategies—tiny drug carriers (nanoparticles), adjusting helpful gut bacteria, and “in-situ vaccines” that activate immune cells inside the tumor—may further boost results. Advanced radiation methods (for example, highly focused stereotactic treatments or proton therapy) aim to protect healthy tissues while keeping the immune signal strong. Because not every patient benefits, researchers are developing simple tests in blood and tumor samples to predict who is most likely to respond. In the future, tools such as artificial intelligence could help match the right dose, field, and timing to each person. Turning this rare effect into a dependable option could open new paths for people with advanced cancers.
Keywords
Introduction
In recent years, significant advances have been achieved in treating solid tumors through surgical interventions, targeted therapies, and systemic treatments. Despite these advances, patients with advanced, metastatic, or treatment-resistant tumors continue to face high mortality and limited therapeutic efficacy. Activating and amplifying systemic antitumor immune responses have therefore emerged as critical strategies to overcome these therapeutic limitations. Radiotherapy (RT), a well-established and widely applied treatment modality, has long been considered a primary means of achieving local tumor control. Nonetheless, accumulating evidence indicates that RT not only exerts direct cytotoxic effects on tumor cells within the irradiated field but also elicits systemic immune responses by inducing immunogenic cell death (ICD), facilitating the release of tumor antigens, and activating innate and adaptive immune pathways. This systemic phenomenon, wherein RT exerts effects on distant, non-irradiated lesions, is referred to as the “abscopal effect,” a term first introduced by Mole in 1953. 1 Indeed, between 1969 and 2014, only 46 rigorously documented abscopal responses were reported, a scarcity that sustained early doubts about the phenomenon’s reproducibility.1 –3 Although traditionally rare following conventional RT, the incidence of the abscopal effect has increased notably with the advent of immunotherapies. RT–immunotherapy combinations have consistently increased the frequency of systemic immune responses, thereby expanding the role of RT from local tumor control to a systemic therapeutic approach. 1 However, abscopal response rates with radio-immunotherapy span a broad range—from single-digit incidence in some series to ≈50% in highly selected cohorts—largely reflecting non-uniform response definitions and marked heterogeneity in study design, tumor histology, and RT-immune checkpoint inhibitor (ICI) sequencing.4 –6 Emerging evidence suggests that the occurrence of the abscopal effect is a hallmark of RT-induced systemic immune activation. Mechanistically, it involves DAMP-mediated activation of antigen-presenting cells (APCs), the mobilization and infiltration of T cells and natural killer (NK) cells, and the remodeling of immune-checkpoint pathways. 7 In multiple tumor types and clinical trials, the abscopal response has been shown to depend on RT-induced antigen release, MHC class I-mediated antigen presentation, CD8+ T-cell activation, and the cooperative amplification of innate immune pathways, including Toll-like receptors and STING signaling. 8 Nevertheless, optimal radiation dose, fractionation, and sequencing for abscopal synergy remain undefined; preclinical work delineates a narrow immunogenic window—single-fraction doses above a TREX1-inducing threshold blunt cGAS–STING signaling.9,10 Moreover, fractionated RT upregulates PD-L1, providing a mechanistic rationale for combining ICIs and for careful scheduling.11,12 Thus, the abscopal effect is increasingly recognized as a critical mediator in radioimmunotherapy, marking a pivotal extension of radiotherapy from local tumor control to systemic therapeutic strategies.
The occurrence of abscopal effects following conventional RT alone remains rare. However, clinical and preclinical studies have consistently shown that combining RT with immunotherapy, especially ICIs, increases both the frequency and extent of abscopal responses. Multiple case reports have documented regression or even complete disappearance of distant, non-irradiated lesions in immunotherapy-responsive tumors such as melanoma, lung cancer, and renal cell carcinoma.13 –15 A recent retrospective analysis investigating the combination of Gamma Knife radiosurgery with ICIs for brain metastases further suggested that RT may promote spontaneous tumor regression at unirradiated sites through systemic immune activation, thereby improving patient survival outcomes. 16 Notably, even in tumors traditionally considered immunologically “cold,” such as pancreatic cancer, esophageal cancer, prostate cancer, and colorectal cancer, appropriately designed RT-immunotherapy strategies have demonstrated the potential to induce systemic immune responses.17 –19 This provides a potential strategy for converting immunologically “cold” tumors into “hot” phenotypes. Despite encouraging reports, abscopal responses remain infrequent; in immunosuppressive TMEs, RT can recruit regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), thereby attenuating systemic antitumor immunity.20 –22 Even so, when RT is combined with appropriate immunomodulators and delivered with defined dose-fractionation/sequence, abscopal regression at unirradiated sites has been documented. For instance, a prospective study in metastatic esophageal squamous cell carcinoma refractory to multiple lines reported that stereotactic body radiotherapy (SBRT) combined with thymosin α1 achieved an (residual cancer burden class 0 or 1; pathologic complete response [RCB 0] or minimal residual disease [RCB 1]) rate of 45.2% and was associated with prolonged distant progression-free survival (PFS), indicating that even highly immunosuppressive tumors can mount systemic antitumor responses when treated with RT plus immune modulators. 17 Moreover, a phase II study in immunotherapy-refractory hepatocellular carcinoma (HCC) suggested that SBRT combined with a PD-1 antibody and anti-angiogenic agents may be a feasible strategy to induce abscopal effects, underscoring the need for prospective validation of abscopal endpoints. 18 Similarly, a recent preclinical study in melanoma models introduced the concept of “boron neutron immunotherapy” (B-NIT), where the integration of boron neutron capture therapy (BNCT) with PD-1 blockade elicited potent CD8+ T-cell-driven abscopal effects in tumors beyond the reach of conventional radiation, providing a potential approach to convert immunotherapy-resistant tumors into immunologically active states. 23
“Immuno-radiotherapy” (iRT) integrates the antigen exposure and immunogenic microenvironment triggered by RT with the immune checkpoint blockade and systemic clonal expansion stimulated by immunotherapy. It has emerged as one of the most actively explored combinatorial strategies in the treatment of solid tumors. RT reshapes the tumor microenvironment (TME) by triggering DNA damage–induced activation of the cGAS–STING pathway and DAMP-mediated immunogenic signaling, thereby enhancing tumor antigenicity, upregulating MHC molecules, and facilitating immune cell infiltration to overcome local immune evasion. Conversely, immunotherapy counteracts the negative immune feedback induced by RT, such as the enrichment of PD-L1, Tregs, and MDSCs, thereby synergistically driving a durable systemic immune response.24 –26 Together, RT and immunotherapy establish a coordinated immune regulatory loop, wherein RT acts to “accelerate” and immunotherapy to “release the brakes,” providing a critical mechanistic foundation for the induction of abscopal effects.
In recent years, the maturation of advanced radiotherapy technologies, including proton therapy, SBRT, FLASH radiotherapy, and spatially fractionated radiotherapy, 27 along with the integration of emerging strategies such as nanotechnology, gene delivery, personalized immune profiling, and AI-assisted radiotherapy planning,28,29 has progressively driven iRT toward greater precision, personalization, and systematization. Concurrently, multiple clinical trials have incorporated iRT into phase II and III study designs across a range of solid tumors, including gastric, lung, breast, liver, and colorectal cancers,30,31 aiming to validate its efficacy in inducing abscopal responses, improving biomarker-based predictive capabilities, and enhancing patient survival outcomes. However, whether these innovations meaningfully enhance abscopal efficacy in patients remains unproven; ongoing trials must determine if proton or FLASH RT—and other novel combinations—increase distant (out-of-field) regression rates beyond conventional regimens.32 –34
Accordingly, we center on clinical evidence in radio-immunotherapy, using only essential mechanistic context, and provide a critical appraisal of controversies, cross-study inconsistencies, and the translational limits of preclinical models.6,35 We define the scope to patient-level, out-of-field responses (“abscopal,” immune Response Evaluation Criteria in Solid Tumors (iRECIST) where available), and standardize terminology to improve cross-study comparability. Mechanistic sections are limited to pathways that directly explain clinical observations (antigen release/MHC-I, cGAS–STING, PD-L1-mediated adaptive resistance) and to key immune populations (Tregs, MDSCs, dendritic cells (DC), macrophages, NK cells). We summarize dose-fractionation and sequencing considerations, biomarker-guided stratification, and the status of emerging modalities (e.g., proton/FLASH), indicating where evidence is sufficient versus pending,4,9,11,36,37 and we outline pragmatic priorities for future trials. Our objective is a concise, clinically oriented framework linking RT parameters, IO timing, and measurable out-of-field responses to guide protocol design and translation.
Molecular mechanisms underlying radiotherapy-induced immune activation
RT can promote the release of DAMPs and tumor-associated antigens (TAAs) from irradiated tumor cells, thereby activating innate and adaptive immune responses. 38 This process involves multiple layers of molecular mechanisms, including DNA damage–sensing pathways, the induction of ICD, and the recruitment and activation of immune cells.
DNA damage and activation of the cGAS–STING pathway
High-dose ionizing radiation directly induces DNA double-strand breaks in tumor cells. Cytosolic and mitochondrial DNA (mtDNA), released upon irradiation, are sensed by the cGAS–STING pathway, which subsequently induces type I interferon production.39,40 For example, blockade of CD73 in tumor cells increases extracellular ATP, enhances RT-induced STING pathway activation, and amplifies type I interferon signaling. This promotes DC activation, T-cell priming, and both local and abscopal antitumor effects. 39 Similarly, strategies that promote mtDNA release—such as cisplatin-induced necroptosis—significantly enhance STING activation and interferon (IFN)-β production, leading to improved antigen cross-presentation and CD8+ T-cell activity. 41 Chen et al. 42 further showed that reduced target volume irradiation D (Reduced Target Radiotherapy) can enhance antigen presentation and T-cell priming by promoting DNA damage and DAMP release, effectively inducing abscopal immune effects without compromising local tumor control. Additionally, a bacterial nanoplatform (VNP@TBTP-Au) designed by Duo et al. 29 was shown to synergize with RT, amplifying DNA damage and reactive oxygen species (ROS) production, thereby activating the cGAS–STING pathway, promoting type I IFN responses, and significantly enhancing CD8+ T-cell infiltration and abscopal effects. In the context of proton therapy, combination with the nanoparticle radioenhancer NBTXR3 has been demonstrated to potentiate cGAS–STING-mediated type I IFN signaling, activating effector T cells, inducing systemic immune memory, and thereby improving survival and reducing pulmonary metastasis. 43 Moreover, liposomal doxorubicin (Doxil) enhances mtDNA release and suppresses mitochondrial transcription factor A (TFAM) expression thereby activating cGAS–STING and IFN-I/CXCL10 signaling to potentiate the abscopal efficacy of RT combined with PD-1 blockade. 44 These results further emphasize the pivotal role of the mtDNA–cGAS–STING axis in radiation-induced immune activation and support the translational potential of mtDNA-releasing agents like Doxil in radioimmunotherapy. 44 In HCC models, 16 Gy radiation activated the cGAS–STING pathway through DNA damage, enhancing DC and CD8+ T-cell activation and suppressing distant tumor growth, underscoring its role in systemic immune activation. 45 Similarly, combining radiotherapy with catalytic nanoparticles (DMPtNPS) and the STING agonist cGAMP effectively remodeled the immunosuppressive microenvironment in rectal cancer, enhancing CD8+ T-cell and DC infiltration and inducing abscopal effects via ROS-mediated hypoxia alleviation and cGAS–STING activation. 46 Importantly, recent clinical observations indicate that intratumoral injection of a STING agonist, combined with PD-1 blockade, induced synchronized regression of both injected and distant lesions in a PD-(L)1-refractory Merkel cell carcinoma patient. The response was durable, lasting over 1 year, and was mechanistically associated with STING activation in immune cells and restoration of tumor HLA-I expression. 47 Collectively, these findings reinforce the central role of the cGAS–STING pathway in radiotherapy-induced immune activation and highlight type I interferon signaling as a critical driver of systemic antitumor immunity.
It is noteworthy that certain molecular pathways may modulate the magnitude of STING-mediated immune responses. For instance, PD-L1 overexpression suppresses STING-mediated T-cell activation and impairs immune responses. In lung cancer models, deletion of tumor-intrinsic PD-L1 significantly enhanced RT-induced cGAS–STING activation, while concurrent autophagy inhibition further amplified type I interferon–mediated antitumor immunity. 40 These findings suggest that targeting immunosuppressive signals like PD-L1 after RT may unlock the full potential of STING-driven antitumor immunity. Additionally, in colorectal cancers lacking cGAS/STING signaling, radiotherapy can still induce type I interferon responses through TLR3-mediated sensing of cytosolic double-stranded RNA, thereby upregulating CXCL10, enhancing CD8+ T-cell infiltration, and promoting abscopal antitumor effects. Adeno-associated virus-mediated overexpression of IFN-β further amplified this response, highlighting the TLR3–dsRNA axis as a crucial compensatory mechanism in STING-deficient tumors. 48 The clinical relevance of this pathway has been exemplified in Merkel cell carcinoma, where intratumoral administration of a STING agonist combined with PD-1 blockade not only reactivated interferon signaling within the TME but also restored HLA-I expression on tumor cells, thereby eliciting robust abscopal responses. 47
ICD and antigen presentation mechanisms
ICD refers to a form of cell death that releases or exposes DAMPs, such as extracellular ATP, high-mobility group box 1 (HMGB1), and calreticulin exposure on the cell surface, thereby promoting immune recognition. RT can induce ICD in tumor cells, transforming tumors into an “in situ vaccine” capable of priming antitumor immunity. For instance, microbeam radiation therapy (MRT) activates ICD and enhances antigen presentation, thereby promoting CD8+ T-cell responses, memory formation, and robust abscopal immunity in melanoma models. 49 Emerging radiotherapy technologies, such as ultra-high dose rate FLASH RT and spatially fractionated RT, have also been shown to potentiate ICD while minimizing normal tissue toxicity. 27 DAMPs released during ICD can attract and activate DCs, facilitating antigen uptake and presentation. Intratumoral injection of oxygen-releasing microparticles alleviates hypoxia, synergizes with RT-induced ICD, and enhances DC-mediated cross-presentation, ultimately promoting systemic CD8+ T-cell responses and controlling distant tumors. 50 Consistently, Trappetti et al. 49 demonstrated that MRT enhances MHC-I antigen presentation and CD8+ T-cell infiltration, eliciting abscopal responses dependent on coordinated activation of cGAS–STING and CD28/CD80 costimulatory pathways, as well as augmented DC and memory T-cell function. In breast cancer models, disulfiram (DSF) combined with copper (Cu) ions was shown to augment RT-induced oxidative stress, significantly increasing the release of ICD markers and promoting CD8+ T cell and DC infiltration while reducing Tregs and MDSCs. 51 Recently, Wang et al. 46 developed a DMPtNPS@cGAMP nanoparticle system that enhanced RT-induced ICD signals (CRT exposure, ATP, and HMGB1 release) and activated DCs and CD8+ T cells via the STING pathway, leading to robust abscopal responses. Similarly, Li et al. 52 constructed a multifunctional QD-Cat-RGD nanoprobe that amplified RT-induced ICD, significantly promoted DC activation and CD8+ T-cell infiltration, and facilitated immune clearance of distant tumors when combined with immunotherapy. Darmon et al. 53 further demonstrated that NBTXR3 nanoparticles, when combined with RT, markedly enhanced ICD responses and diversified the tumor immunopeptidome, promoting CD8+ T-cell-mediated abscopal effects. In addition, Kemmotsu et al. 54 reported that hydrogen peroxide combined with radiotherapy increases ICD, augments HMGB1 release and calreticulin exposure, promotes DC maturation and CD8+/IFN-γ+ T-cell infiltration at distant sites, thereby amplifying abscopal responses; PD-1 blockade further enhances tumor control. Building upon this, Zhou et al. developed a strategy targeting cancer-associated fibroblasts (CAFs) via photodynamic activation of CAF-related ICD, using a TME-responsive FAPα-targeted probe (FMP). Localized photodynamic therapy combined with PD-L1 blockade not only eradicated primary tumors but also significantly inhibited the progression of untreated distant lesions. These effects were attributed to CAF depletion, ICD enhancement, systemic CD8+ T-cell expansion, and reduction of Tregs and MDSCs. 55 Yang et al. developed a metabolic intervention combining ^177Lu-labeled radioactive seed implantation with IDO1 inhibitor-loaded alginate microspheres to induce ICD and promote tumor antigen release. This approach concurrently suppressed IDO1-driven immunosuppression in the TME and synergized with PD-L1 blockade to elicit abscopal responses in non-irradiated lesions. 56 Collectively, these findings highlight ICD as a central mechanism by which radiotherapy initiates systemic antitumor immune responses. Enhancing ICD via pharmacologic or nanotechnologic strategies offers a promising route to augment abscopal responses and improve clinical efficacy. As illustrated in Figure 1, the RT-induced cascade underpins enhanced antigen presentation, T-cell priming, and the initiation of systemic (abscopal) immune activation.

Radiotherapy-induced ICD and systemic antitumor immunity (abscopal effect). RT induces local tumor cell death and immunogenic stress, leading to the release of TAAs and DAMPs (ATP, HMGB1, CRT). RT also upregulates immunostimulatory molecules (e.g., MHC-I, ICAM-1, B7-H3, Fas) on surviving tumor cells. Cytosolic tumor-derived DNA activates the cGAS–STING pathway, promoting IFN-I production and DC maturation. Recruited DCs capture TAAs and traffic to draining lymph nodes for cross-presentation and prime CD8+ T cells. Activated CTLs then infiltrate distant tumors and kill tumor cells via perforin and granzyme—constituting the abscopal effect. RT also induces immunosuppressive ligands (PD-L1, CD47, CTLA-4), which can be counteracted by immune checkpoint blockade to sustain systemic control. Left-to-right flow: local RT-induced immune activation → antigen presentation/T-cell priming → abscopal tumor killing; the bottom insets summarize STING, ICD, and CD8+ effector mechanisms.
The antigens and danger signals released during ICD require efficient antigen presentation to elicit effective T-cell responses. Radiotherapy-induced apoptosis and necrosis in tumor cells release a large quantity of neoantigens. Locally, DCs can capture these antigens and migrate to draining lymph nodes to initiate T-cell priming. In parallel, radiotherapy-induced stress can upregulate the expression of MHC class I molecules and costimulatory molecules such as ICAM-1 and Fas on surviving tumor cells, rendering them more susceptible to cytotoxic T lymphocyte (CTL)-mediated clearance. 57 For example, radiotherapy has been shown to enhance the expression of ICAM-1 and B7-H3 in solid tumors, thereby improving the efficacy of subsequent CAR-T-cell therapies. In bilateral tumor models, radiotherapy combined with B7-H3-targeted CAR-T cells eradicated irradiated tumors and enhanced CAR-T infiltration and cytotoxicity in non-irradiated lesions, underscoring the synergy between RT-induced antigen release and ICD in augmenting systemic adoptive cell therapy responses. 57 Moreover, tumor vaccine strategies incorporating adjuvants can leverage radiotherapy-induced antigen release to further boost immune activation. For instance, an in situ vaccine comprising a TLR9 agonist (CpG) and OX40 costimulatory antibody, when combined with local radiotherapy, reprogrammed the tumor immune microenvironment (TIME) and suppressed non-injected distant tumors, thereby enhancing systemic antitumor immunity. 58
TME remodeling and immune cell recruitment
RT exerts a bidirectional influence on the TME. On one hand, RT enhances the production of chemokines and cytokines, thereby promoting the recruitment of effector immune cells into tumors. On the other hand, RT may upregulate immunosuppressive molecules, leading to the accumulation of suppressive cell populations. For instance, Han et al. 59 showed that single high-dose irradiation in a breast cancer model not only increased intratumoral CD8+ T cells but also elevated infiltration by Tregs, MDSCs, and M2-polarized tumor-associated macrophages (TAMs). Such accumulation of immunosuppressive cells remains a major limiting factor for the induction of abscopal effects. To validate this, a dual PI3Kγ/δ inhibitor was used to selectively deplete MDSCs and Tregs, which significantly increased effector T-cell infiltration and enhanced RT-induced abscopal responses. 59 In a bilateral bladder cancer model, Rompré-Brodeur et al. 60 showed that RT plus PD-L1 blockade downregulated CCL22/IL-13 and upregulated CXCL9/granzyme B (GZMB), remodeling the TME, enhancing CD8+ T-cell activity, and producing robust abscopal clearance. Liu et al. confirmed in a dual-tumor model the critical role of TME immune-cell remodeling in abscopal responses. Removal of tumor-draining lymph nodes (TDLNs) significantly impaired CD8+ T-cell and M1-macrophage infiltration/activation (Granzyme B+, IFN-γ+) in both irradiated and distant tumors and reduced the M1/M2 ratio, compromising abscopal control. 61 Similarly, Chang et al. 62 showed that a PI3Kαδ inhibitor reduced Tregs/MDSCs in a triple-negative breast cancer model and, with RT plus PD-1 blockade, significantly enhanced CD8+ T-cell effector function, promoting immune clearance of distant non-irradiated tumors. CAFs are a major obstacle to abscopal activation. Zhou et al. 55 showed that FAPα-targeted ablation with the FMP plus PD-L1 blockade markedly enhanced CD8+-T-cell-mediated suppression of distant tumors. Consistently, Hou et al. 26 showed that RT induced tumor-cell CXCL10, recruiting MDSCs to distant non-irradiated sites and promoting immunosuppression and potential metastasis; PD-L1/CXCL10 blockade reversed these effects and improved distant control. Recently, in CT26 colorectal cancer, local RT plus the dual PI3Kδ/γ inhibitor BR101801 reduced Treg and M2-like macrophage infiltration, maintained a favorable effector CD8+-cell ratio, and upregulated IFN-γ and CXCL9/10, thereby enhancing local and distant antitumor responses. 63 Lan et al. developed bintrafusp alfa (BA), a bifunctional fusion protein targeting PD‑L1 and TGF‑β. In multiple “cold” tumor models, BA reversed RT‑induced immunosuppression, promoted CD8+ T‑cell and dendritic‑cell trafficking to primary and distant tumors, and reduced Treg/MDSC infiltration, remodeling the TME. Mechanistically, BA targets PD‑L1+/TGF‑β+ endothelium and M2‑like fibroblasts and synergizes with RT‑induced type I IFN/STING signaling to convert “cold” to “hot” tumors and induce abscopal effects. 64 Thus, although RT stimulates immune activation, concomitant immunosuppressive circuits within the TME may constrain systemic antitumor immunity and should be co‑targeted.
Overall, RT remodels the TME by simultaneously “releasing the brakes” and “stepping on the accelerator” of antitumor immunity. On the one hand, RT promotes tumor-antigen release and type I interferon production, thereby attracting and activating DCs and T cells, accelerating antitumor immune responses.38,39 On the other hand, RT-induced DNA damage and cellular stress upregulate PD-L1 and recruit suppressive populations (Tregs, MDSCs), thereby applying immunosuppressive “brakes.”26,59 Given that RT alone often fails to fully drive systemic antitumor immunity, combining RT with immunotherapy is theoretically compelling: pharmacologic interventions that release immune suppression (lifting the brakes) and simultaneously boost immune activation (pressing the accelerator) could maximize abscopal effects. Representative molecular pathways mediating RT-induced immune activation and abscopal effects are summarized in Table 1. The balance between these immunostimulatory and immunosuppressive mechanisms is further illustrated in Figure 2. The PEMBRO-RT study demonstrated that SBRT combined with PD-1 blockade enhanced CD103+ CD8+ T-cell infiltration and lymphoid aggregation in non-irradiated lesions relative to PD-1 monotherapy, thereby indicating that SBRT can amplify immune-mediated abscopal effects and remodel the TME. 65 Recent studies showed that incorporating CD122-targeted IL-2 complexes into RT plus PD-1 blockade expanded circulating stem-like CD8+ T cells expressing high CXCR3, facilitating their trafficking to and clearance of distant non-irradiated tumors. 66 Walker et al. 67 reported that the CD122-biased IL-2 agonist NKTR-214 synergized with local RT to enhance CD8+ expansion and effector function at both irradiated and abscopal sites, improving cure rates and overall survival (OS) in multifocal models. In murine melanoma and colorectal models, Onyshchenko et al. 68 demonstrated that hypofractionated RT plus lenalidomide enhanced DC cross-presentation, CD8+ activation, and TA-HEV formation, thereby improving TME remodeling and inducing potent abscopal effects. In pancreatic cancer models, proton therapy synergized with mesothelin-targeted CAR-T by upregulating antigen expression and reshaping immune cell lineages within the TME, expanding CAR-T cells and increasing IFN-γ at non-irradiated sites—features indicative of systemic immune activation. 69 Moreover, non-radiative local interventions have also been shown to stimulate abscopal responses. Recent preclinical studies found that microwave ablation (MWA) increased Th1 cytokines and CD8+ functionality, remodeled the TME, and suppressed untreated tumors, suggesting that thermal ablation may potentiate systemic immunity. 70 Zhang et al. 71 engineered Salmonella strains to secrete nattokinase, degrading the extracellular matrix and suppressing CAF activity, thereby increasing TME permeability and enhancing RT-induced DC/CD8+ infiltration, which augmented abscopal effects and promoted immune-memory formation in colorectal cancer models. Similarly, Lei et al. 72 engineered an M13 bacteriophage–based in situ vaccine that synergized with RT to induce ICD, activate DCs, and reverse local immunosuppression; when combined with PD‑1 blockade, it increased T‑cell infiltration and abscopal effects. Additionally, curcumin has been reported to act as an immunomodulator, enhancing RT‑induced T‑cell activation and inducing pro‑inflammatory cytokines (IL‑1β, IL‑6); in colorectal cancer models, it strengthened abscopal responses and upregulated the T‑cell activation marker OX40. 73 Finally, Fujimoto et al. 23 showed that, in immunotherapy‑resistant melanoma, BNCT combined with PD‑1 blockade activated CD8+ T cells and suppressed both irradiated and distant lesions, providing the first experimental evidence that BNCT can induce abscopal effects.
Representative molecular pathways mediating radiotherapy-induced immune activation and abscopal effects.
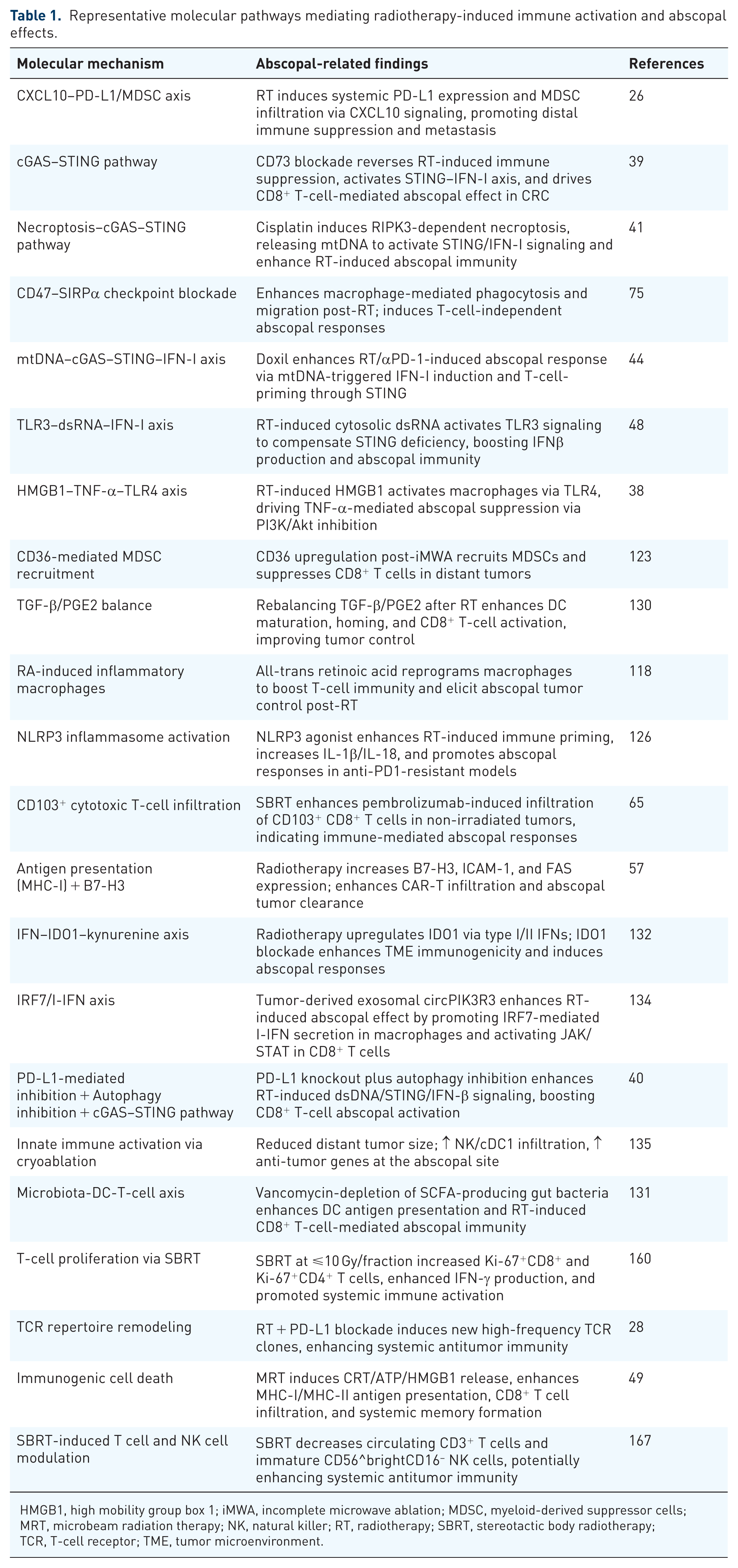
HMGB1, high mobility group box 1; iMWA, incomplete microwave ablation; MDSC, myeloid-derived suppressor cells; MRT, microbeam radiation therapy; NK, natural killer; RT, radiotherapy; SBRT, stereotactic body radiotherapy; TCR, T‑cell receptor; TME, tumor microenvironment.

Dual immunomodulatory effects of radiotherapy on the tumor microenvironment. (a) RT upregulates TGF-β and IL-10, promoting Treg expansion via SMAD2/3, STAT3, and FoxP3, with epigenetic support. (b) RT-conditioned TAMs polarize toward an M2 phenotype via NF-κB/ROS signaling, secreting IL-10, TGF-β, and CCL22, reinforcing immunosuppression. (c) RT enhances DC activation via DAMPs and TAAs; NK-derived chemokines (CCL5, XCL1) further recruit DCs. B7–CD28 and CCR7–CCL19/21 signaling support T-cell priming and DC trafficking. (d) Tumor-derived CCL2 and CSF-1 activate STAT3 and PI3K–AKT pathways in TAMs, promoting M2 polarization and suppressive function. Panels (a) and (b) depict RT-amplified suppressive circuits, whereas (c) and (d) illustrate pro-immune pathways; the net balance governs the likelihood of distant (abscopal) control.
Molecular mechanisms of radiotherapy-induced abscopal effects and clinical translation
Synergistic mechanisms between radiotherapy and immunotherapy
Given the multifaceted impact of RT on the immune system, combining RT with immunotherapy—especially immune checkpoint blockade—is a rational approach to enhance the induction of abscopal effects. In this section, we elucidate the synergistic mechanisms between RT and major checkpoint inhibitors—PD‑1/PD‑L1 and CTLA‑4—and emerging immunomodulatory pathways (e.g., LAG‑3, glucocorticoid-induced tumor necrosis factor receptor (GITR)). Furthermore, we discuss how factors such as radiation dose, fractionation schemes, and treatment sequencing influence the efficacy of these combinatorial strategies.
Synergistic effects with immune checkpoint blockade
The PD-1/PD-L1 axis
The PD-1/PD-L1 pathway represents a major mechanism of tumor immune evasion. RT has been shown to upregulate PD-L1 expression on both tumor cells and tumor-infiltrating immune cells, thereby suppressing T-cell activity. 26 Even in “cold” EGFR‑mutant non-small cell lung cancer (NSCLC) with limited ICI responsiveness, Xia et al. 74 showed that RT activated cGAS–STING and increased systemic CD8+ infiltration, thereby sensitizing tumors to PD‑L1 blockade and inducing robust abscopal responses. Accordingly, combining RT with PD-1/PD-L1 inhibitors can mutually amplify antitumor immunity. Notably, Lan et al. showed that adding BA (PD‑L1/TGF‑β) to RT significantly remodeled the TME, improved local control, and induced abscopal effects in untreated lung metastases. Mechanistically, effects correlated with increased CD8+/NK infiltration, reduced TAMs/MDSCs, and suppression of RT‑induced fibrosis and immunosuppression. 64 Beyond T-cell-mediated effects, CD47—a myeloid immune checkpoint—has also emerged as a synergistic target. CD47 blockade plus RT induced macrophage‑driven abscopal responses in small‑cell lung cancer models, offering a T‑cell-independent immunologic strategy. 75 Multiple preclinical studies have consistently validated this synergy. In murine melanoma models, RT alone yielded limited abscopal effects, whereas adding anti-PD‑1 significantly improved control of non‑irradiated tumors; this benefit was CD8+ T‑cell dependent. Importantly, this mechanism has also been corroborated in clinical settings. In a retrospective study by Deguchi et al., 76 dogs with pulmonary metastatic oral malignant melanoma receiving hypofractionated RT followed by PD‑L1 blockade achieved a 55.6% objective response at metastatic sites, significantly higher than the 10% in non‑irradiated controls, with prolonged survival. Park et al., 24 using bilateral B16‑OVA melanoma and RENCA models, found that PD‑1 deletion or anti‑PD‑1 therapy significantly augmented SABR (15 Gy)-induced abscopal responses and prolonged survival, with enrichment of CD11a^high^ PD‑1+ CD8+ T cells closely associated with tumor regression. 24 Similarly, Regenold et al. 77 showed that thermosensitive liposomal vinorelbine plus mild hyperthermia achieved only limited local control, whereas robust abscopal effects emerged only with the addition of anti-PD‑1, underscoring the central role of ICIs in systemic immune activation. Emerging strategies further implicate modulation of the gut–immune axis. A prospective study showed that low‑dose intestinal irradiation plus PD‑L1 blockade augmented T‑cell immunity at distant sites and improved tumor control, supporting a gut–immune contribution to RT-ICI synergy. 78 Yoo et al. 45 established a dual‑tumor HCC model in which RT plus PD‑1 blockade increased CD8+ T‑cell infiltration, activated DCs, and polarized TAMs toward M1 in non‑irradiated tumors, yielding durable systemic control and survival benefits. In a bladder cancer model, Rompré-Brodeur et al. 60 reported that RT plus PD-L1 blockade delayed growth of irradiated and non-irradiated tumors, prolonged survival, and remodeled the TME (↑CXCL9/GZMA/GZMB; ↓CCL22/IL-13). In PD‑1-resistant NSCLC models, Chen et al. 79 reported that adding SHP2 inhibition to RT and PD‑L1 blockade enhanced abscopal responses via M1 macrophage polarization, augmented CD8+ T‑cell activity, and Treg depletion, thereby reversing immune resistance. The SWORD study demonstrated that SBRT plus PD‑1 blockade and GM‑CSF achieved a 30.6% RCB 0/1 abscopal response rate in advanced NSCLC, implicating T‑follicular helper (Tfh) cells and IL‑21 as key mediators. 30 He et al. 80 further augmented these effects by targeting M2 macrophages with a STAT6 antisense oligonucleotide combined with hypofractionated RT and PD‑1 blockade, significantly improving primary and distant tumor control and survival, and linking efficacy to TGF‑β suppression, CD8+ T‑cell rejuvenation, and TAM reprogramming. In breast cancer models, low‑dose total‑body irradiation (L‑TBI) combined with high‑dose hypofractionated RT and PD‑1 blockade synergistically enhanced CD8+ T‑cell and dendritic‑cell infiltration, limited MDSC accumulation, and amplified systemic immune activation. 81 In 4T1 triple-negative breast cancer, PD-1 blockade augmented RT-induced “vaccination effects” by promoting tumor-specific T-cell clonal expansion and RCB 0/1 responses in pulmonary metastases. 82 Similarly, local injection of Bacillus Calmette–Guérin (BCG) with low‑dose RT (LDRT) significantly boosted abscopal control and CD8+ T‑cell infiltration, supporting the potential of non‑ICI immunostimulatory strategies. 83 Clinical case reports provide further corroborative evidence. For instance, Postow et al. 84 reported a melanoma patient who progressed on CTLA‑4 blockade (ipilimumab) but showed regression of both irradiated and non‑irradiated lesions after RT, with increased NY‑ESO‑1 antibody titers, more ICOS+ T cells, and fewer MDSCs—providing mechanistic insight into RT-ICI synergy. More recently, a PD‑1-refractory metastatic melanoma patient exhibited regression of non‑irradiated metastases after low‑fraction RT, indicating that RT can re‑ignite systemic immunity even in checkpoint‑refractory settings. 13 Collectively, these data indicate that PD-1/PD-L1 blockade relieves RT-induced T-cell inhibition (“releasing the brakes”), allowing RT-released antigens and IFN signals to drive CTL-mediated clearance.24,26
The CTLA-4 pathway
CTLA‑4 blockade acts early in T‑cell priming, attenuating DC-mediated coinhibition and depleting Tregs, thereby augmenting antitumor T‑cell responses. RT promotes the presentation of newly released tumor antigens, providing a strong rationale for its synergy with anti-CTLA-4 therapies. 85 Zhou et al. reviewed key studies (e.g., CheckMate‑204, KEYNOTE‑189) and highlighted that combining SRS or WBRT with PD‑1/CTLA‑4 blockade activated intracranial immune microenvironments, increased blood–brain barrier permeability, and drove local T‑cell clonal expansion. Importantly, “concurrent” administration was shown to induce more robust intracranial abscopal-like responses and significantly improve local control and OS compared to sequential strategies. 86 In a landmark study, Formenti et al. 85 showed that CTLA‑4 blockade alone offered limited benefit in metastatic NSCLC, whereas adding thoracic RT produced shrinkage or stabilization of non‑irradiated metastases, supporting synergy between RT‑induced immune priming and CTLA‑4 blockade. Similarly, in murine pancreatic cancer models, high‑dose RT combined with anti-CTLA‑4 therapy achieved superior systemic antitumor efficacy relative to either monotherapy. This combination reduced intratumoral Treg infiltration, increased CD8+ CTL density, delayed growth of distant lesions, and prolonged OS. 87 Notably, both single‑fraction high‑dose RT (e.g., 20 Gy × 1) and hypofractionated moderate‑dose RT (e.g., 8 Gy × 3) synergized effectively with CTLA‑4 blockade in preclinical models. 87 These findings suggest that, within an appropriate dose range, RT‑induced antigen release and immune activation can cooperate with CTLA‑4 blockade to promote systemic and abscopal antitumor responses.
Emerging immune checkpoints (eg., LAG-3 and GITR)
Beyond the classical immune checkpoints PD-1 and CTLA-4, emerging checkpoints and costimulatory pathways are increasingly being incorporated into radioimmunotherapy strategies. Recent work by Zhang et al. showed that cotargeting CD39 and VISTA reversed RT‑induced immunosuppression by limiting terminal CD8+ T‑cell exhaustion and suppressing immunosuppressive tumor-associated neutrophils (TANs) and monocytic MDSCs (M-MDSCs). Notably, CD39 blockade plus PD‑1 inhibition with RT significantly increased T‑cell infiltration, fostered immune memory, elicited robust abscopal responses, and prolonged survival. 88 LAG-3, a critical marker of T-cell exhaustion, has also emerged as a promising target. A recent case report showed that nivolumab (anti-PD-1) plus relatlimab (anti-LAG-3), given with liver SBRT, controlled the irradiated nasal mucosal melanoma lesion and induced regression of distant bone metastases, suggesting that LAG-3 blockade may further potentiate RT-induced abscopal effects. 89 GITR, a costimulatory receptor highly expressed on Tregs within tumors, has been shown to be a valuable target. GITR agonist antibodies not only enhance T-cell effector functions but also selectively deplete intratumoral Tregs, thereby reversing local immunosuppression. GITR is a costimulatory receptor expressed on T cells, predominantly enriched on Tregs within the TME. Agonistic antibodies targeting GITR have been shown to not only enhance effector T-cell function but also selectively deplete intratumoral Tregs, thereby reversing local immunosuppression. 90 Notably, although radiotherapy can promote antigen release and T-cell infiltration, it may simultaneously lead to the enrichment of Tregs, thereby dampening abscopal immune responses. Schoenhals et al. 90 showed, in a PD‑1-resistant NSCLC model, that RT plus PD‑1 blockade and a GITR agonist markedly depleted intratumoral Tregs, enhanced CD4+/CD8+ memory T‑cell responses, and induced abscopal effects in non‑irradiated tumors, with durable remissions in some mice. Additionally, localized co-delivery of IL-2 with CTLA-4 blockade can amplify RT-induced abscopal effects. Au et al. 91 developed a tumor-injected nanocomposite tumor-injected nanocomposite (TIN) that, in response to post-RT hypoxia, released IL-2, α-CTLA-4 antibodies, and poly(I:C), thereby expanding CD8+ T cells and facilitating clearance of distant tumors, representing an in situ vaccination strategy. Similarly, in a Lewis lung carcinoma “cold‑tumor” model, L19–IL2 plus RT and PD‑L1 blockade achieved a 38% complete‑response rate and effective abscopal clearance, supporting triplet regimens (“RT + immune accelerators + checkpoint blockade”) for activating systemic immunity in resistant tumors. 92 OX40 costimulation represents another promising target for enhancing radiotherapy-induced immune responses. In a murine breast cancer model, the combination of an OX40 agonist, PD‑1 blockade, and RT significantly reduced primary and distant tumor burden and was associated with a higher CD8+/Treg ratio, less CD8+‑T‑cell exhaustion, greater dendritic‑cell activation, and higher IL‑2/IFN‑γ levels. 93 Moreover, Martin et al. 94 showed that RT‑induced upregulation of 4‑1BB synergized with anti‑4‑1BB to activate B cells and CD4+ T cells, amplify CD8+‑mediated antitumor responses, and suppress distant tumor growth. Building on these findings, Jiang et al. showed, in a liver-metastasis model of Lewis lung carcinoma, that triple therapy (RT + PD-1 blockade + anlotinib) significantly inhibited both primary and distant subcutaneous tumors, prolonged survival, increased infiltration of CD8+/IFN-γ+ T cells and DCs, and reduced polymorphonuclear (PMN)-MDSCs. RNA-seq revealed activation of the PPAR pathway, implicating metabolic reprogramming as a contributor to enhanced immune responses, particularly in immunologically “cold” liver metastases. 95 Carlson et al. 96 further proposed that supplementing local RT combined with immunotherapy (e.g., in situ tumor immunization (IT-IC)) with low-dose irradiation (2–6 Gy) or targeted radionuclide therapy delivered to all known lesions could enhance systemic antitumor immunity by reinforcing immune memory at the primary site and suppressing non-irradiated lesions. This study highlights that, for multifocal metastatic disease, combining “whole‑lesion irradiation” with “localized immune activation” may be an optimal strategy to maximize abscopal responses. In parallel, incorporating CD122‑biased IL‑2 complexes into RT plus PD‑1 blockade regimens markedly expanded CXCR3+ stem‑like CD8+ T cells, thereby enhancing abscopal effects and providing a reservoir of effector T cells for subsequent adoptive cell therapy. 66 Similarly, Walker et al. 67 confirmed that NKTR‑214 combined with RT significantly improved abscopal tumor‑clearance rates (up to 86%) and activated robust CD8+ T‑cell-mediated systemic responses in CT26 and other models, highlighting CD122‑targeted IL‑2 agonists as potent synergists of abscopal effects. 67 In addition, Zhou et al. reported that methylglyoxal (MG), a gut microbiota-derived metabolite, functions as a novel radiosensitizer by increasing ROS, inducing ER stress and DNA damage, and thereby activating the cGAS–STING pathway. In a CT26 bilateral‑tumor model, MG combined with RT and PD‑1 blockade achieved complete remission of all irradiated tumors and induced abscopal responses in approximately 50% of non‑irradiated lesions, supporting the clinical translational potential of MG‑augmented radioimmunotherapy. 19 Collectively, these findings suggest that pairing RT with dual checkpoint blockade and/or costimulatory agonists may yield greater synergy than targeting PD‑1 or CTLA‑4 alone. For instance, in preclinical models, concurrent PD‑1 and CD73 blockade—targeting a key immunometabolic checkpoint—achieved greater tumor control than PD‑1 inhibition alone. 97 Clinically, dual blockade of PD-1 and CTLA-4 has already demonstrated improved response rates in several refractory tumors, and integrating radiotherapy into this strategy holds promise for further amplifying therapeutic outcomes.
Challenges in optimizing dose fractionation and treatment sequencing
Although synergy between RT and immunotherapy is widely recognized, optimizing dose-fractionation and the timing of immunotherapeutic interventions remains a central challenge. Regarding dose fractionation, different regimens can exert distinct effects on the immune system. Several studies support hypofractionated high‑dose RT (⩾8 Gy per fraction over a few fractions), which more reliably induces ICD and STING‑mediated type I interferon responses, thereby increasing immunogenicity. For instance, in a murine pancreatic cancer model, Mills et al. 98 found that SBRT, relative to conventional fractionated RT, synergized with IL-12 gene therapy to remodel the TIME—promoting M1 macrophage polarization and CTL infiltration—resulting in eradication of both primary and metastatic tumors. However, high-dose RT may also cause substantial lymphocyte depletion and normal tissue toxicity. In contrast, LDRT can preserve and activate local immune cells. Interestingly, a retrospective analysis in breast cancer patients suggested that for individuals who had already received high-dose RT and immunotherapy, the addition of LDRT to untreated metastases significantly increased the incidence of abscopal responses: 58% of LDRT-irradiated lesions exhibited partial or complete responses compared to only 18% of unirradiated lesions. 99 Moreover, Chen et al. 78 showed across multi-cohort retrospective and preclinical studies that low-dose non-targeted gut irradiation (1–3 Gy) significantly enhanced systemic responses to PD-L1 blockade, mechanistically associated with upregulation of mregDCs, gut-microbiota remodeling—particularly enrichment of Christensenella species—and activation of systemic CD8+ T cells. These findings suggest that low-dose radiation may enhance systemic T-cell responses by inducing low-level antigen release and regulatory cytokine production. In bilateral tumor models, the impact of dose regimen on abscopal effects was evident: single-fraction high-dose RT (20 Gy × 1) induced stronger abscopal tumor suppression than moderate-dose fractionation (8 Gy × 3). 100 In murine bilateral-tumor models, intentionally heterogeneous intratumoral dosing—high-dose “peaks” (~16 Gy) interdigitated with low-dose “valleys” (~2 Gy)—achieved superior control of unirradiated lesions when combined with PD-1 blockade, and efficacy was further augmented by CXCR2 inhibition.101,102 Mechanistically, peak regions elicited ICD and cGAS–STING-dependent type-I-interferon signaling, whereas valley regions—a hallmark of spatially fractionated/lattice RT—spared APCs and intratumoral CD8+ T cells, thereby preserving in situ priming.9,102 Consistent with an adaptive-immune requirement, T-cell depletion abrogated these effects, indicating that spatial dose heterogeneity operated as an in situ vaccination strategy rather than a purely cytotoxic schema.24,102 Mechanistically, spatially fractionated radiotherapy (SFRT) imposes a peak–valley geometry in which ablative peaks (~15–20 Gy) are interleaved with low-dose valleys (~0.5–2 Gy). 103 Peaks elicit profound DNA damage, DAMP release, and cGAS–STING-driven type-I IFN, whereas valleys spare APC niches and intratumoral lymphocytes, preserving in situ priming otherwise lost with uniform fields.101,103 In vivo, partial-tumor irradiation can selectively activate STING and—despite reduced target coverage—achieve tumor control comparable to whole-tumor irradiation in immunocompetent hosts, but not when adaptive immunity is absent. 104 Early clinical implementations (lattice RT; SBRT-PATHY) have reported non-targeted/abscopal regressions with acceptable toxicity, supporting spatial dose heterogeneity as a clinically actionable immunomodulator.105,106 FLASH-RT, by reducing normal-tissue injury and treatment-related lymphopenia, may provide complementary immune preservation to spatial patterning.27,107 In support of this, Zhang et al. 108 used the N@VP nanoplatform to evaluate LDRT (2 Gy × 3 fractions) and found that it better preserved immune cell function and remodeled the TME compared to higher-dose regimens, offering a translational model for optimizing radio-immunotherapy using low-dose strategies. Collectively, these results indicate that there is currently no universally optimal radiation protocol for combining with immunotherapy; dose optimization must consider tumor type, anatomical location, and the specific mechanisms of the partnered immunotherapy.
The timing of ICI administration relative to RT critically affects the magnitude of the abscopal effect. Theoretically, RT-induced tumor antigens and type I interferon signals require a certain interval to initiate T-cell responses, suggesting that administering ICIs after RT may be more conducive to expanding effector T-cell clones. However, experimental findings on this issue remain inconsistent. In a well-controlled murine study, Wei et al. demonstrated that administration of anti-PD-1 antibodies after RT significantly enhanced inhibition of distant tumor growth. Mechanistically, the benefit reflected expansion of polyfunctional CD8+ T cells and reduced exhaustion following RT-induced activation; by contrast, anti-PD-1 given before RT precipitated premature exhaustion and blunted systemic responses. 109 In contrast, a study conducted in an osteosarcoma model reported that administration of anti-PD-L1 antibodies either before or after radiotherapy resulted in comparable improvements in tumor control compared to monotherapy, suggesting that the sequence of administration might not always be critical. 110 Clinically, various strategies have been adopted. Some trials have utilized concurrent administration of radiotherapy and ICIs, whereas others have introduced radiotherapy after several cycles of immunotherapy to augment systemic responses. For instance, in patients with advanced melanoma resistant to PD‑1 blockade, low‑dose, fractionated RT has been used to reinvigorate systemic antitumor immunity. Funck‑Brentano et al. 13 reported that ≈35% of these patients had regression or shrinkage of non‑irradiated lesions after low‑dose irradiation. Overall, the optimal timing for integrating ICIs with radiotherapy appears to be disease-specific, and further prospective trials are warranted to systematically compare different sequencing strategies.
Beyond dose and timing considerations, the extent of the radiation field also significantly influences immune synergy. Traditionally, to ensure tumor control, radiotherapy fields have included generous safety margins, but this approach may inadvertently damage surrounding lymphocyte-rich regions critical for immune priming. Emerging studies have proposed the concept of “field contraction” radiotherapy, wherein imaging-guided techniques are used to precisely restrict radiation fields, thereby preserving adjacent normal lymphoid structures. Prospective animal studies demonstrated that precise delivery of immune stimulators embedded in biodegradable polymers, combined with localized low-dose irradiation, significantly enhanced CD8+ T-cell infiltration and systemic immune activation at distant tumor sites, particularly in immunologically “cold” tumors. 111 Similarly, Liu et al. found that the integrity of TDLNs was indispensable for inducing abscopal responses with combined radioimmunotherapy. Ablation of TDLNs markedly impaired CD8+ T-cell infiltration, decreased the M1/M2 macrophage ratio, and ultimately attenuated distant tumor control. 61 Chen et al. 42 further demonstrated in murine models that minimizing radiation field size significantly reduced peripheral lymphocyte depletion, preserved tumor-adjacent immune infiltrates, and amplified systemic antitumor immunity induced by radiotherapy. Mechanistically, this strategy restricted high-level DNA damage to tumors while preserving lymphocyte viability, enabling robust activation of the cGAS-STING pathway and effector T-cell responses. Clinically, convergent observations suggest comparable effects. In the IMM‑101 trial in unresectable pancreatic cancer, investigators combined SBRT with a heat‑inactivated Mycobacterium obuense vaccine to enhance immunogenicity and used image‑guided SBRT to minimize irradiation of normal tissues. Increases in ICOS+ and Ki‑67+ T cells without a significant rise in TIM‑3/LAG‑3 were observed, consistent with cooperative immune activation. 112 Looking forward, the integration of advanced technologies, such as MRI-guided adaptive radiotherapy and real-time immune monitoring, holds promise for further refining the optimal combination of dose, field, and sequencing parameters, thereby maximizing the induction of abscopal effects while maintaining acceptable toxicity profiles and enabling the development of personalized radioimmunotherapy protocols.
Preclinical studies and case summaries of radiotherapy-induced abscopal effects
Role of key immune cell populations in the abscopal response
A variety of preclinical models has been leveraged to dissect the immunological mechanisms driving the abscopal effect. Among these, the bilateral flank tumor model—with localized irradiation applied to a single lesion—remains the benchmark platform for evaluating systemic tumor responses. This model consistently demonstrates that regression of non-irradiated lesions is closely linked to dynamic remodeling of the tumor–immune microenvironment. Multiple immune cell subsets, including CD8+ T cells, Tregs, MDSCs, DCs, macrophages, and NK cells, have been implicated as critical regulators of this phenomenon.
CD8+ T cells
CD8+ T cells are pivotal effectors mediating radiotherapy-induced antitumor immunity, particularly in the context of the abscopal effect. Emerging clinical strategies, such as combining radiotherapy with engineered T-cell therapies (e.g., CAR-T), have significantly advanced systemic tumor control. For instance, Ma et al. 113 demonstrated in a pancreatic cancer model that Claudin18.2 (CLDN18.2)-specific CAR-T cells, combined with local 4 Gy irradiation, elicited robust systemic immune responses, evidenced by complete regression of irradiated tumors and significant suppression of non-irradiated lesions. Radiotherapy-induced tumor cell apoptosis promoted the expansion of both CAR-T cells and endogenous CD8+ T cells, while increased chemokine expression (e.g., CCL2, CXCL9) in distant tumors enhanced T-cell recruitment and infiltration, thus facilitating systemic antitumor immunity. These findings highlight that radiotherapy not only initiates antigen release but also reshapes chemokine-driven immune cell trafficking, thereby enhancing the systemic efficacy of adoptive cell therapies such as CAR-T, offering a novel approach to precisely elicit abscopal responses. Furthermore, combining radiotherapy with ICIs (e.g., PD-L1 blockade) or immunocytokines (e.g., L19–IL2) effectively activated CD8+ memory T cells in immunologically “cold” tumors, enhancing distant tumor control and long-term immune memory formation. 92 Moreover, Duo et al. 29 demonstrated that the combination of VNP@TBTP-Au nanoparticles and radiotherapy significantly induced ICD and activated CD8+ T cells, resulting in robust inhibition of distant tumors, suggesting that CD8+-mediated abscopal responses can be potentiated through nanomaterial delivery systems synergized with immune pathways. Traditional chemotherapeutic agents have also been shown to augment abscopal effects via immune mechanisms. Cisplatin-induced RIPK3-dependent necroptosis triggered mtDNA release, cGAS–STING activation, type I interferon secretion, and enhanced DC-mediated cross-presentation, substantially increasing CD8+ T-cell activation and abscopal tumor control. 41 Radiotherapy significantly enhanced CD8+ T-cell activity in brain metastases, and when combined with precisely timed PD-1 blockade, synergistically augmented abscopal responses despite central immune privilege. 86 Similarly, administration of PD-1 blockade following radiotherapy amplified systemic CD8+ T-cell-mediated antitumor responses. 109 Beyond CAR-T strategies, Fujimoto et al. proposed and validated a novel radiotherapy–immunotherapy paradigm termed B-NIT in a melanoma model. B-NIT, combining BNCT with PD-1 blockade, activated CD8+ effector memory and early activated T cells, significantly suppressing distant tumor growth. Mechanistically, this response involved the release of DAMPs, expansion of TILs, and antigen spreading. 23
Regulatory T cells
Radiotherapy-induced immune responses are often accompanied by the accumulation of Tregs, which represents a major mechanism of tumor-mediated immune resistance. Tregs express immunosuppressive molecules (e.g., CTLA-4, FoxP3, CD73), inhibiting effector T-cell proliferation and secreting suppressive cytokines including IL-10 and TGF-β. Multiple preclinical studies have focused on strategies to attenuate Treg-mediated immunosuppression in order to amplify the abscopal effect. Immune checkpoint blockade (e.g., anti-CTLA-4) effectively reduces intratumoral Treg proportions, enhancing CTL infiltration and suppressing both irradiated and distant tumor growth in combination with radiotherapy. 87 Alternatively, costimulatory receptor activation (e.g., GITR agonism) can impair Treg function and induce apoptosis. Combination with radiotherapy reduces Treg accumulation, promotes effector T-cell activation, and significantly enhances distant tumor control. 90 Similarly, sequential radiotherapy and OX40 agonist administration significantly enhances local and abscopal tumor control by promoting CD4+ and CD8+ T-cell expansion, CD103+ DC recruitment, and systemic antitumor immunity, notably in PD-1-resistant tumors. 114 In triple-negative breast cancer models, Han et al. 93 demonstrated that triple combination therapy involving an OX40 agonist, RT, and PD-1 blockade increased the CD8+/Treg ratio by more than threefold and reduced the proportion of exhausted PD-1+ CD8+ T cells in distant tumors, thereby potentiating the abscopal effect. In colorectal cancer bilateral tumor models, BR101801, a PI3Kδ/γ dual inhibitor, combined with RT, was shown to suppress Tregs and enhance CD8+ T-cell function, successfully inducing abscopal responses and demonstrating the effectiveness of Treg-targeting strategies even in immunologically “cold” tumors. 63 Additionally, radiotherapy combined with tumor-targeted IL-2 immunocytokines (e.g., L19–IL2) substantially controls distant tumors and promotes durable CD8+ memory T-cell responses, crucial for sustained abscopal immunity. 115 This approach has already entered early-phase clinical trials in non-small cell lung cancer (NCT02086721), supporting the translational potential of these findings. In addition to direct T-cell activation or Treg suppression, inducing direct apoptosis of tumor cells through radiotherapy has also been shown to significantly enhance abscopal responses. Recent studies by Xu et al. 116 in melanoma models demonstrated that the combination of RT with a CD95 agonist antibody significantly promoted apoptosis in both primary and distant tumors, increased CTL and DC infiltration, and robustly enhanced the induction of abscopal effects.
In addition to the aforementioned immunotherapeutic strategies, modulation of the survival environment of Tregs represents another promising approach. Wu et al. 117 reported that the IL-33/ST2 signaling axis plays a pivotal role in maintaining Treg proliferation and function. Both radiotherapy and MWA were shown to release IL-33, which in turn activated the ST2 receptor on Tregs, promoting their expansion and immunosuppressive capabilities. In a murine HCC model, MWA alone failed to induce abscopal effects and instead increased the proportion of Tregs within residual tumors through IL-33/ST2-mediated signaling. However, when ST2 knockout mice or ST2-blocking antibodies were employed, a significant reduction in Treg accumulation was observed, accompanied by enhanced CD8+ T-cell effector function in distant tumors post-MWA, ultimately resulting in the emergence of a pronounced abscopal effect. 117 These findings suggest that targeting inflammation-associated pathways, such as IL-33–ST2 signaling activated by tumor injury, may represent a viable strategy to regulate Treg-mediated immunosuppression and critically influence the success of abscopal immune responses.
Small-molecule agents, such as all-trans retinoic acid (ATRA), induce inflammatory macrophages, promote effector T-cell proliferation, and impair Treg function, thus augmenting radiotherapy-induced abscopal effects. 118 In a breast cancer model, the combination of ATRA and radiotherapy significantly increased the CD8+ T/Treg ratio, and the addition of PD-L1 blockade further enhanced the abscopal effect. 118 Another notable example involves histone modification inhibitors: Kim et al. 119 found that BRD4 inhibition not only reduced PD-L1 and HIF-1α expression in tumor cells but also suppressed the infiltration of immunosuppressive cells, including Tregs, thereby synergizing with radiotherapy to improve CTL infiltration and cytotoxic function in murine breast cancer models. Clinical studies also support Treg involvement in abscopal responses, as elevated Treg activity correlated with poorer outcomes in breast cancer patients undergoing SBRT. Thus, combining radiotherapy with Treg-targeted strategies may potentiate clinical abscopal effects. 120 Overall, multi-targeted interventions against Tregs—whether through antibody-mediated depletion (e.g., anti-CTLA-4, GITR agonists, OX40 agonists) or modulation of cytokine environments and gene expression (e.g., IL-33/ST2 blockade, ATRA, BRD4 inhibition)—have been consistently shown to release the immunosuppressive “brakes” imposed by Tregs, thereby synergizing with radiotherapy-induced antigen release to amplify systemic antitumor immune responses.
Myeloid-derived suppressor cells
MDSCs constitute another class of immunosuppressive cells frequently enriched following RT, comprising two primary subsets: PMN-MDSCs and M-MDSCs populations. MDSCs suppress T-cell proliferation through mechanisms involving the production of arginase, iNOS, and various immunosuppressive mediators, thereby facilitating tumor immune evasion. Multiple studies have demonstrated that RT increases the proportion of MDSCs both in tumors and in peripheral blood circulation, which may attenuate systemic antitumor immunity and hinder abscopal responses.26,59 Therefore, strategies aimed at reducing the abundance or function of MDSCs hold promise for improving the systemic efficacy of radiotherapy. Preclinical studies indicate that PI3Kγ/δ inhibition reduces RT-induced MDSC infiltration, restores T-cell function, and enhances abscopal effects. 59 Similarly, in a humanized cold immune phenotype urothelial carcinoma model, Yamamoto et al. 121 reported that RT alone had limited effects on controlling distant tumors and led to a marked accumulation of PMN-MDSCs in non-irradiated sites. However, when MDSC-targeted therapies—such as chemokine receptor blockade or recruitment inhibition—were combined with RT, significant suppression of distant tumor growth was observed. Notably, improved control occurred independently of significant CD8+ T-cell infiltration, suggesting MDSC depletion alone sufficiently alleviates immunosuppression to amplify abscopal effects. These findings collectively emphasize that, particularly in immunologically “cold” tumors, the key to improving systemic therapeutic outcomes may lie less in boosting effector T-cell quantity and more in alleviating the suppressive influence exerted by MDSCs.
In addition to direct depletion strategies, multiple approaches have been proposed to target MDSCs. One strategy involves inhibiting tumor-derived myelopoietic factors. VEGFR2 blockade has been shown to reduce tumor angiogenesis and GM-CSF expression, thereby decreasing MDSC and Treg recruitment. Concurrently, anti-VEGFR2 treatment enhanced DC activation and CCL5-mediated CD8+ T-cell migration into tumors, ultimately augmenting the abscopal effects of RT combined with anti-PD-1 therapy in a HCC model. 122 These findings suggest that VEGF pathway inhibition may partially “normalize” tumor vasculature and alleviate immunosuppression, creating a more favorable environment for effector T-cell infiltration. Another promising approach involves metabolic targeting of MDSCs. Liu et al. 123 reported that incomplete microwave ablation induced upregulation of CD36, leading to the accumulation of MDSCs in distant non-ablated tumors and impairment of T-cell function, thereby promoting an immunosuppressive abscopal effect. Notably, administration of a CD36 inhibitor reversed this process, reducing suppressive MDSC populations and restoring T-cell activity at distant sites. Given the association of CD36 with lipid metabolism, these findings highlight metabolic pathways as potential intervention targets for attenuating the immunosuppressive function of MDSCs.
Moreover, modulation of RT parameters itself can influence the behavior of MDSCs. Clinically, adding low-dose irradiation to high-dose RT significantly increased abscopal responses, likely by reprogramming the TME and reducing pro-tumorigenic myeloid cell activity. 99 In a murine model, Bergeron et al. 101 demonstrated that applying spatially heterogeneous irradiation (combining low-dose and high-dose regions within the same tumor) alongside CXCR2 chemokine receptor blockade (to inhibit neutrophil and MDSC intratumoral trafficking) resulted in superior systemic antitumor responses and prolonged survival compared to uniform high-dose irradiation. Collectively, these findings reinforce that MDSCs are critical contributors to immune negative feedback following RT. Preclinical studies consistently show that pharmacologic or strategic interventions targeting MDSCs can substantially enhance abscopal effects.59,121,122 Moving forward, integrating MDSC-targeting strategies into clinical practice represents a significant opportunity to enhance the translational potential of abscopal immunotherapy.
Dendritic cells and antigen presentation
DCs serve as pivotal mediators in the initiation of primary T-cell responses and are critically involved in the induction of abscopal effects. The abundant tumor antigens released following RT must be captured, processed, and presented by DCs to T cells within draining lymph nodes to elicit a systemic immune response. Emerging evidence indicates that RT-induced immunogenicity—particularly via STING—relies on BATF3+ classical type I DCs (cDC1s), which specialize in cross-presenting antigens to CD8+ T cells. Intratumoral injection of STING agonist BO-112 combined with RT significantly enhanced CD8+ T-cell infiltration. This effect depended on type I interferon receptors and BATF3-dependent DCs. 124 These findings suggest that the combination of RT with additional STING activation can enhance DC-mediated antigen presentation, thereby facilitating robust abscopal responses. In an HCC abscopal model, Park et al. 125 showed that RT upregulated type I-interferon expression and increased CD11c+ DC accumulation in TDLNs, leading to recruitment and activation of CD8+ T cells at distant, non-irradiated sites. Consistent with these observations, a clinical study in Merkel cell carcinoma showed that intratumoral administration of a STING agonist activated tumor‑infiltrating DCs and enhanced HLA‑I-mediated antigen presentation, thereby increasing infiltration of tumor‑specific CD8+ T cells and inducing abscopal effects; these findings underscore the central role of DC activation in systemic immune responses following RT. 47
To further enhance DC-mediated immunogenicity, several strategies have been explored to directly activate or recruit DCs into the TME. For instance, in the PD-1-resistant 344SQ-R lung adenocarcinoma model, intratumoral administration of an NLRP3 inflammasome agonist in combination with stereotactic body radiotherapy (12 Gy × 3 fractions) enhanced DC activation and cross-presentation, resulting in potent abscopal tumor regression despite immunotherapy resistance. 126 These findings suggest that direct DC activation may represent a critical strategy to overcome radioimmunotherapy resistance. Similarly, Onyshchenko et al., 68 using a bilateral tumor model, demonstrated that lenalidomide enhanced type I interferon-dependent DC cross-presentation, upregulated costimulatory molecules (CD70, CD83, CD86), and induced tumor-associated high endothelial venule(s) (TA-HEV/TA-HEVs) formation, significantly amplifying CD8+ T-cell-mediated abscopal effects and durable immune memory following low-fraction RT. In breast cancer models, a Fe12-POM-based radiocatalytic system induced ICD and activated DC function, thereby synergizing with PD-1 blockade to suppress distant tumors, further underscoring the central role of antigen presentation in abscopal responses. 127 Moreover, Gao et al. developed an implantable hydrogel “cell factory” that sustained CCL21 release to recruit DCs and T cells. When implanted in primary tumors and combined with localized RT, the CCL21‑DCs@hydrogel enhanced intratumoral DC infiltration and activation and suppressed metastasis and recurrence, 128 thereby creating an in situ “DC drainage center” that facilitated immune priming after RT-induced antigen release. In addition, Pang et al. 129 demonstrated that nanoparticle-mediated activation of the TLR4 pathway significantly enhanced DC maturation and antigen presentation, driving effector T-cell expansion and amplifying systemic abscopal effects, further confirming the pivotal role of DCs in RT-induced immune responses. Recent studies report that RT can disrupt the TGF-β/PGE2 axis, impairing DC migration and function and limiting antigen presentation and abscopal efficacy. Timely TGF-β blockade restored PGE2-driven immunostimulatory effects, enhancing DC maturation, trafficking, and systemic activation. 130 Single-cell transcriptomics further revealed that late-phase RT impairs cDC1 maturation while promoting expansion of cDC2-derived mDCs expressing higher levels of immunosuppressive molecules (e.g., IL-1β, PD-L1), suggesting that RT may bias DC lineage fate and dampen antigen presentation and T-cell activation. 88 Additionally, gut-microbiota modulation influences DC function. Uribe-Herranz et al. 131 showed that oral vancomycin depleted butyrate-producing bacteria (e.g., Clostridia), relieving inhibitory effects on DCs and enhancing RT-induced antigen cross-presentation, boosting IFN-γ-dependent CD8+ T-cell responses, and improving clearance of distant tumors. Furthermore, local DSF/Cu injection plus 12 Gy RT in breast cancer models enhanced ICD, promoted DAMP release and subsequent DC activation, thereby amplifying antigen-presentation capacity. 51 Similarly, engineered M13 bacteriophages activated DCs by displaying CD40 ligand and delivering GM-CSF; intratumoral injection at irradiated sites served as an in situ vaccine that enhanced local and systemic antitumor responses and synergized with PD-1 blockade to induce robust abscopal effects. 72
In addition to directly enhancing DC activation, targeting immunometabolic pathways that impair DC function has emerged as a complementary strategy to augment antigen presentation. IDO1, an enzyme expressed by tumor and immune cells, catabolizes tryptophan into immunosuppressive kynurenine, which suppresses the function of both DCs and T cells. Recently, a radiolabeled microsphere co-delivering an IDO1 inhibitor with radiotherapy and immune checkpoint blockade significantly reprogrammed the TME in preclinical HCC models. This strategy restored DC function and significantly enhanced abscopal responses. 56 In CRC models, radiotherapy upregulated IDO1 via interferon signaling in colorectal cancer, contributing to tumor radioresistance. IDO1 inhibition (e.g., epacadostat) or genetic deletion reversed immunosuppression, increasing radiosensitivity, Th1 cytokines (IFN-γ, TNF-α), effector T-cell infiltration, and inducing abscopal tumor regression. 132 Collectively, these findings highlight IDO1 as a critical negative regulator of RT-induced immunity; its inhibition may enhance DC-mediated T-cell priming and systemic antitumor responses.
Importantly, the status of TDLNs—the primary site for antigen presentation and T-cell activation by DCs—plays a decisive role in shaping abscopal responses. Preclinical studies have shown that successful radioimmunotherapy-induced abscopal effects correlate with increased CD8+ T-cell infiltration and an elevated M1/M2 macrophage ratio in TDLNs. 61 Conversely, removing TDLNs or disrupting DC–T-cell interactions significantly impaired systemic tumor control. These findings underscore the necessity of preserving TDLN integrity during RT-ICI combination therapy to maintain effective antigen priming and immune initiation. In clinical radiotherapy planning, unnecessary irradiation of functional TDLNs should be avoided to prevent collateral damage to key immune structures. This rationale parallels the concepts behind field-sparing techniques such as involved-node RT and SBRT-PATHY, which aim to preserve immune-competent zones, ensuring sufficient “ammunition and manpower” for systemic immune engagement.
Macrophages, NK cells, and other innate immune components
Innate immune cells are indispensable participants in the induction of RT-mediated abscopal effects, with TAMs and NK cells being the most extensively studied subsets. TAMs exhibit remarkable plasticity, with M1-like macrophages exerting pro-inflammatory and antitumor effects, while M2-like macrophages support tumor progression and immune suppression. RT has been shown to influence the polarization state of TAMs. For example, using antisense oligonucleotides to inhibit STAT6—a transcription factor driving M2 polarization—reprogrammed TAMs toward an M1 phenotype following radiotherapy, marked by increased IL-12 and reduced TGF-β production, thereby fostering an immune-permissive TME. 80 Moreover, combining RT, MerTK inhibition, and anti-PD-1 therapy synergistically activates macrophages, NK cells, and CD8+ tissue-resident memory T cells, inducing robust abscopal tumor regression in NSCLC and pancreatic cancer models. 133 In the same NSCLC setting, a triple regimen involving STAT6 ASO, RT, and anti-PD-1 not only reduced M2 TAM and Treg populations but also enhanced Th1-type immune responses. As a result, significant suppression of both primary and non-irradiated distant tumors was achieved. 80 Collectively, these findings highlight macrophage reprogramming as critical for creating an immune-permissive TME, effectively promoting T-cell activation and abscopal responses.
Macrophages can directly mediate abscopal effects by secreting inflammatory cytokines. In a murine breast cancer model, radiotherapy-induced release of HMGB1 activated TLR4-dependent M1 macrophage polarization, resulting in elevated TNF-α production. This cytokine subsequently suppressed proliferation and migration of non-irradiated tumor cells via the PI3K–p110γ signaling cascade, thereby contributing to abscopal tumor regression. 38 Notably, macrophage depletion or TNF-α neutralization abolished this effect, confirming the macrophage-cytokine axis in abscopal responses. These findings suggest that enhancing TAM pro-inflammatory activity, via TLR4 agonists or GM-CSF, may amplify abscopal effects during radiotherapy. In a similar context, SBRT combined with local IL-12 delivery in pancreatic cancer models significantly activated intratumoral IFN-γ signaling, shifting TAMs toward an M1 phenotype. This, in turn, initiated a systemic CD8+ T-cell response that eradicated non-irradiated liver metastases and established long-term immune memory. 98 This study clearly illustrated the central role of TAMs in the multistep radioimmunologic cascade: from radiotherapy-induced macrophage polarization to subsequent T-cell activation and abscopal tumor regression. Building upon this, in PDAC models, SBRT was shown to upregulate CD73 on TAMs and myeloid cells, fostering an immunosuppressive TME. A triple combination of SBRT, anti-CD73, and anti-PD-L1 antibodies significantly downregulated IL-6, VEGF, and MIP-1α/β, while enhancing TAM-mediated antigen presentation, pro-inflammatory signaling, and long-term immune memory, leading to immune-mediated clearance of distant tumors. 97 Furthermore, radiotherapy-induced inflammatory chemokines, such as CSF1, recruited macrophages, and CD47 blockade activated TAMs to mediate T-cell-independent abscopal antitumor effects, suggesting the CD47–SIRPα axis as a potential therapeutic target beyond the PD-1/CTLA-4 pathways. 75 Most recently, Wang et al. 134 demonstrated in a bilateral melanoma model that radiotherapy triggers tumor‑derived circPIK3R3‑containing exosomes, which sequester miR‑872‑3p to derepress IRF7, amplify type I interferon signaling, and polarize macrophages toward an M1 phenotype, thereby priming JAK1–STAT1 activation in CD8+ T cells and enhancing clearance of non‑irradiated pulmonary metastases.
As innate lymphocytes, NK cells possess unique advantages in combating metastatic tumors. Although NK cells exert cytotoxicity independently of antigen presentation, their antitumor efficacy can be compromised under immunosuppressive conditions, allowing tumor cells to evade immune surveillance. Cryoablation of primary breast tumors significantly upregulated immune-related gene expression and remodeled the TIME in distant untreated lesions. This effect involved increased recruitment of NK cells and type 1 conventional DCs dendritic cells (cDC1s) delaying abscopal tumor growth. 135 These findings suggest that local tumor ablation or radiotherapy, if capable of activating NK cells, may contribute to the induction of abscopal effects. In radiotherapy models, L-TBI has been shown to activate NK cells. When combined with local radiotherapy and PD-1 blockade in a breast cancer model, L-TBI led to increased infiltration of NK cells and CD8+ T cells in distant, non-irradiated tumors, significantly enhancing the abscopal response. 81 These results support the concept that NK cells serve an auxiliary “ice‑breaking” function early in systemic immune activation, delivering immediate cytotoxicity that facilitates T‑cell priming and the initiation of RT‑induced systemic immunity. Furthermore, in the LuM-1 murine colorectal cancer model, radiotherapy plus metformin improved local tumor control and markedly suppressed the growth of distant, non-irradiated pulmonary metastases. This effect coincided with increased proportions of splenic CD8+ T cells and CD335+ NK cells, elevated IFN-γ, and accumulation of activated NK cells within lung metastases. 136 Together, these observations nominate augmentation of cytotoxic effector compartments—particularly NK cells—as a rational strategy to potentiate abscopal responses.
In addition to macrophages and NK cells, neutrophils and other myeloid-derived cells also play important roles in the modulation of abscopal responses. A specific neutrophil subset, TANs, can promote tumorigenesis under certain conditions, thereby impairing immune-mediated tumor clearance. Blockade of neutrophil infiltration via CXCR2 antagonism has been shown to enhance the immunological efficacy of radiotherapy. 101 Conversely, neutrophils may also act as facilitators of antitumor immunity. In clinical observations of radiotherapy in head and neck cancer, patients who exhibited abscopal responses typically showed a post-treatment decrease in the peripheral neutrophil-to-lymphocyte ratio (NLR), suggesting a systemic immune shift toward lymphocyte predominance. 137 These findings underscore the context-dependent dual role of neutrophils, which warrants further investigation based on TME characteristics. B cells have also emerged as contributors to certain forms of abscopal effects. In a striking observation, Martin et al. found that the abscopal efficacy of anti-4-1BB therapy depended on the cooperation between B cells and T cells. Upon combination of radiotherapy with a 4-1BB agonist, B cells were found to accumulate within the TME and potentially function as accessory APCs or organizers of tertiary lymphoid structures, thereby supporting T-cell-mediated tumor clearance. 94 These results highlight a previously underappreciated role of B cells in enhancing the quality of T cell responses in the context of radioimmunotherapy.
Collectively, preclinical evidence indicates that abscopal effects arise from coordinated actions of multiple immune cell subsets. RT‑induced DAMPs recruit innate immune cells—including macrophages, neutrophils, and NK cells—to initiate the inflammatory cascade and clear necrotic debris. These innate responses facilitate antigen uptake and presentation by DCs, which in turn activate and expand CTLs that traffic to and eliminate distant metastases. Throughout this process, immunosuppressive cells such as Tregs and MDSCs antagonize effective immune responses. Notably, genetic knockouts, pharmacologic inhibitors, and antibody‑mediated depletion have consistently enhanced RT‑mediated abscopal antitumor effects across murine models by modulating the abundance or activity of these immune‑cell subsets.59,80,90,121 These mechanistic insights provide a valuable framework for the rational design of combinatorial therapeutic strategies. Representative preclinical and clinical combination regimens that have successfully induced abscopal responses across various tumor models are summarized in Table 2. In the following section, we will discuss recent clinical advances in radioimmunotherapy and how these preclinical findings are being translated into clinical benefit for patients.
Synergistic radio-immunotherapy regimens inducing abscopal responses in preclinical and clinical models.

CLDN18.2, Claudin18.2, DC, dendritic cells; ICI, immune checkpoint inhibitors; LDRT, low-dose RT; MDSC, myeloid-derived suppressor cells; MSS, microsatellite-stable; PFS, progression-free survival; RT, radiotherapy; SABR, stereotactic ablative body radiotherapy; SBRT, stereotactic body radiotherapy; SIRT, selective internal radiation therapy; STR, spontaneous tumor regression; TAM, tumor-associated macrophages; TANs, tumor-associated neutrophils; TCR, T‑cell receptor; Tfh, T‑follicular helper; TME, tumor microenvironment; TRM, tissue-resident memory T cells.
CR, complete response; CRC, colorectal cancer; GM-CSF, granulocyte-macrophage colony-stimulating factor; MPR, major pathological response; ORR, objective response rate; PR, partial response; SRS, stereotactic radiosurgery; WBRT, whole brain radiation therapy.
Clinical advances and optimization strategies in radioimmunotherapy
In recent years, the combination of radiotherapy and immunotherapy has achieved several breakthrough advances in clinical settings. Multiple tumor types—including NSCLC, melanoma, renal cell carcinoma, HCC, and colorectal cancer—have been investigated to evaluate the efficacy and safety of this combined strategy. Concurrently, clinical trials are actively exploring optimization pathways, such as dose fractionation schemes, sequencing of immunotherapy, and incorporation of third-agent immunomodulators, with the goal of improving abscopal response rates and patient survival. The following section provides an overview of clinical evidence and strategic developments.
Notwithstanding robust murine data, clinically demonstrable abscopal responses remain uncommon, reflecting host-, tumor-, and treatment-parameter differences between mice and humans. In patients—especially those with immune-excluded or immunosuppressive microenvironments—CAF-rich stroma and the accumulation of MDSCs and Tregs impede effector trafficking and blunt RT-elicited systemic priming.138,139 Moreover, regimens that are permissive in mice (e.g., high single fractions or irradiation of all lesions) are clinically constrained by normal-tissue tolerance: large or elective nodal fields can deplete lymphocytes and sterilize TDLNs, diminishing antigen presentation.140,141 Conversely, very high per-fraction doses can induce the DNA exonuclease TREX1, degrading cytosolic DNA and extinguishing cGAS–STING/type-I-IFN signaling, thereby abrogating immunogenicity.9,142 These realities support trial designs that maximize safe coverage of dominant disease while sparing TDLNs, use immunogenic yet lymphocyte-sparing fractionation, and align checkpoint blockade within the RT-primed window; by contrast, subablative partial coverage (e.g., CHEERS) may be insufficient to overcome systemic suppression.
Clinical efficacy of radiotherapy combined with immunotherapy
In unresectable stage III NSCLC, PACIFIC established durvalumab consolidation after concurrent chemoradiotherapy as standard of care, improving PFS and OS with durable long-term benefit (sustained at 5 years). 143 In metastatic NSCLC, the randomized phase II PEMBRO-RT trial showed that SBRT to a single lesion before pembrolizumab increased objective responses versus pembrolizumab alone without excess grade ⩾3 toxicity, supporting an ablative-priming strategy. Beyond PEMBRO-RT, multiple phase II studies are testing SBRT plus ICIs—concurrently or sequentially—to elicit systemic (abscopal) immunity. In the peer-reviewed SWORD phase II trial, 30 30 patients were enrolled with metastatic NSCLC who received SBRT in combination with the anti-PD-1 antibody sintilimab and GM-CSF. The triple combination yielded enhanced objective response rates in both primary and metastatic lesions, with over 40% of non-irradiated tumors demonstrating disease control, and 1-year OS notably improved. 30 These results suggest that adding an immune adjuvant such as GM-CSF, which promotes APC activation, may further enhance the systemic antitumor response induced by radiotherapy and PD-1 blockade. In the ComIT-1 trial, SBRT plus the anti-PD-L1 antibody atezolizumab resulted in an RCB 0/1 disease control rate in previously treated advanced NSCLC. 144 Reductions in circulating tumor DNA (ctDNA) correlated with therapeutic efficacy, and no uncontrollable toxicity was observed, indicating that the regimen was effective and well tolerated. Van der Woude et al. 65 further observed that in certain NSCLC patients treated with SBRT and ICIs, radiographic abscopal responses were not always apparent; however, immune biopsies of non-irradiated lesions revealed favorable remodeling, including increased infiltration of CD8+ T cells and other immune constituents. This suggests that radioimmunotherapy can induce systemic immune reshaping even without measurable tumor shrinkage. Formenti et al. provided the first clinical evidence that local radiotherapy combined with anti-CTLA-4 therapy could induce abscopal tumor regression in metastatic NSCLC. The response was associated with upregulation of IFN-β and dynamic changes in T-cell clonality, thereby supporting the underlying mechanistic synergy. 85 Additionally, several case reports have documented SBRT-induced abscopal responses in NSCLC patients receiving nivolumab. 14 The ImmunoSABR prospective randomized trial further demonstrated that the combination of SABR with the vascular-targeted immunocytokine L19–IL2 induced immunologic clearance of non-irradiated metastases in patients with stage IV NSCLC, accompanied by improved survival outcomes. 31 These findings highlight the potential of multi-modal immunotherapeutic strategies to extend the clinical utility of radiotherapy in metastatic lung cancer. Notably, benefit has not been universal. In the randomized phase II CHEERS trial, adding limited-site SBRT (3 × 8 Gy to ⩽3 lesions) to PD-1/PD-L1 inhibitors across multiple tumor types did not improve PFS or OS versus immunotherapy alone. 145 These hypothesis-generating data suggest that subablative, partial irradiation may be insufficient for durable systemic effects, and that broader lesion coverage and/or ablative dosing might be required to realize abscopal synergy.
To sharpen clinical relevance and enable cross-trial comparison, we specify whether prospective studies prespecified out-of-field regression and which standardized endpoints they used. In NSCLC, phase II designs such as SWORD prospectively captured abscopal shrinkage of non-irradiated lesions, while ComIT-1 linked rapid ctDNA declines to regression at unirradiated sites.144,146 Across tumor types, recent protocols increasingly apply iRECIST for distant-lesion assessment and incorporate molecular readouts (e.g., ctDNA kinetics) to detect early systemic benefit. 147 These harmonized endpoints support causal inference that radiotherapy can convert local injury into measurable system-wide control.
In HR-positive/HER2-negative breast cancer (NCT03804944), a randomized, four-arm phase II neoadjuvant trial randomizes postmenopausal patients—after 4 months of letrozole—to focal hypofractionated radiotherapy (8 Gy × 3) alone or combined with pembrolizumab, recombinant FLT3 ligand (CDX-301), or both; the RT-only arm serves as the control, the single-agent arms isolate the independent effects of PD-1 blockade and FLT3L, and the triplet arm tests additivity/synergy (ClinicalTrials.gov identifier: NCT03804944).148,149 This design aligns with the mechanistic rationale that RT releases tumor antigens and can induce PD-L1, FLT3L expands cross-presenting cDC1, and PD-1 blockade reverses T-cell exhaustion—together enabling in situ vaccination.11,150,151
In immune-sensitive tumors such as melanoma and renal cell carcinoma, RT has been shown to augment clinical responses to immune checkpoint inhibition. Funck-Brentano et al. evaluated the effect of delayed low-dose radiotherapy in patients with advanced melanoma who had progressed on anti-PD-1 monotherapy. Following low‑dose, fractionated radiotherapy (3 Gy × 5) to multiple metastatic sites, approximately one‑third of patients exhibited regression or complete remission of non‑irradiated lesions, consistent with a classical abscopal effect. 13 Importantly, no excess immune‑related adverse events were observed, indicating that RT may reinvigorate systemic immunity in ICI‑refractory disease with an acceptable safety profile. Mechanistically, Trappetti et al. 49 used MRT in the B16‑F10 melanoma model to systematically characterize abscopal responses, showing that MRT significantly increased MHC‑I/II antigen presentation and drove robust CD8+ T‑cell infiltration; importantly, CD8+‑cell depletion abrogated efficacy. Notably, MRT also activated the cGAS–STING pathway and the CD28/CD80 costimulatory axis, promoting memory CD8+‑T‑cell differentiation and initiating vaccine‑like systemic priming in TDLNs, culminating in durable abscopal antitumor immunity and immune memory formation. Clinically, ImmunoSABR demonstrated that SABR plus the tumor-targeting immunocytokine L19–IL2 activated CD8+ T cells and induced regression at non-irradiated sites in NSCLC. Findings were attributed to synergy between IL-2-driven immune activation and RT-mediated antigen release. 31 In HCC, Park et al. 125 established a preclinical abscopal model showing that a single 8 Gy localized irradiation induced CD8+ T‑cell infiltration and activation in distant, non‑irradiated tumors; anti-PD‑L1 therapy markedly prolonged this response, highlighting that sustained abscopal responses depend on timely immunologic intervention to overcome post‑radiation immune tolerance. In renal cell carcinoma, Margulis et al. conducted a phase II trial in which patients with RCC and inferior vena cava tumor thrombus received neoadjuvant SABR (59 Gy/3 fractions) before radical nephrectomy. The preliminary results indicated that the regimen was both safe and feasible. Postoperative biomarker analyses showed reduced Ki-67 expression and elevated PD-L1 levels in some patients, suggestive of immune activation. 152 Although the study focused on surgical feasibility and local control, the findings raise the possibility that high-dose RT promotes immune-mediated clearance of micrometastases and could synergize with postoperative immunotherapy. Additionally, a case report described advanced gastric cancer with brain metastases treated with whole‑brain RT plus PD‑1 blockade. Not only did the intracranial lesions respond favorably, but distant extracranial metastases also exhibited marked regression, resulting in durable survival. 15 This case illustrates that, in traditionally immunologically “cold” tumors (e.g., gastric and colorectal cancers), RT may help overcome constraints imposed by the blood–brain barrier and an immunosuppressive TME, thereby restoring responsiveness to immune checkpoint blockade.
In gynecologic malignancies, Kao et al. reported a case of platinum-refractory recurrent endometrial clear cell carcinoma (ECCC) in which disease progression occurred during treatment with pembrolizumab. Adding CyberKnife RT to control local disease induced a robust abscopal effect, yielding complete remission. Notably, the patient remained disease-free for over 3 years after treatment discontinuation. 153 This case highlights the clinical relevance of abscopal responses even in immunologically “cold” tumors such as ECCC, suggesting that the combination of immune checkpoint inhibition and radiotherapy can elicit durable systemic immune activation.
Gastrointestinal malignancies with poor prognoses—notably HCC and pancreatic cancer—are being investigated for radioimmunotherapy strategies. Zhao et al. conducted a prospective, single-center phase II trial (SACTION01) testing neoadjuvant SBRT plus tislelizumab (anti-PD-1) and chemotherapy in unresectable NSCLC. Although conducted in lung cancer, this study provides proof‑of‑concept for triple‑modality therapy in locally advanced solid tumors: 76% achieved major pathological response, including a substantial proportion with complete pathological remission. 154 In extensive-stage small-cell lung cancer, real-world data have also shown that adding thoracic RT to PD-L1 inhibitors plus chemotherapy significantly prolongs both PFS and OS, further supporting the synergistic potential of RT-ICI combinations in systemic disease control. 155 These results suggest that for selected locally advanced unresectable tumors—including subsets of HCC and pancreatic cancer—RT combined with immunotherapy and chemotherapy may enable surgical conversion or durable disease control. In advanced pancreatic cancer, promising preclinical results by Mills et al. 98 have prompted clinical testing of SBRT plus IL‑12-based immunotherapy. In early clinical reports, patients receiving this combination exhibited notable T-cell infiltration within the TME, and several cases demonstrated prolonged disease stabilization. 156 Although clinical data remain limited, these findings offer insight into overcoming the intrinsic immune resistance of this “cold” tumor type.
In CRC, most patients with advanced disease exhibit microsatellite-stable (MSS) status, which is associated with resistance to ICI monotherapy. Consequently, radioimmunotherapy strategies are particularly attractive in this setting. A multicenter phase II trial (NCT03104439) evaluated SBRT plus atezolizumab in metastatic CRC, including immune correlates. Levy et al. reported disease control in a subset of refractory MSS CRC after SBRT plus PD‑L1 blockade. This was accompanied by significant remodeling of peripheral and intratumoral immune profiles. 157 In-depth immunologic analyses revealed that baseline intratumoral expression of PD-L1 and IRF1 was higher in responders. Furthermore, treatment induced upregulation of key chemokines and cytotoxic genes—such as CCL19, CXCL9, and GZMB—within the TME. 157 Collectively, these data suggest that in an immunologically “hotter” subset of MSS CRC, RT may facilitate immune‑cell recruitment. Despite transient lymphodepletion caused by radiotherapy, the remaining immune cells may still be functionally reactivated and redirected toward tumor infiltration, thereby partially overcoming primary resistance to ICIs. In another study involving patients with hepatic metastases from MSS CRC, the FFCD 1709-SIRTCI phase II trial investigated the combination of selective internal radiation therapy (SIRT, using ^90Y-labeled microspheres targeting liver metastases) with chemotherapy (XELOX), bevacizumab, and atezolizumab. The five-drug combination showed manageable safety and early signals of efficacy. 158 Notably, in some patients who had previously progressed on chemotherapy and targeted therapy, liver lesions regressed following the addition of SIRT and immunotherapy. This outcome suggests that multimodal strategies incorporating locoregional radiotherapy may sensitize MSS metastatic CRC to ICIs. While the long-term survival benefits of such regimens require further investigation, this study reflects a growing trend toward multi-pronged therapeutic approaches for refractory CRC liver metastases. Overall, although MSS CRC remains a prototypical immune-resistant tumor, ongoing clinical trials of radioimmunotherapy are beginning to demonstrate encouraging signals of efficacy. However, further data are needed to optimize patient selection and refine combination strategies in this challenging population.
Optimization of dose, timing, and combination strategies in radioimmunotherapy
Clinical experience and trial evidence have provided important insights into the optimization of radioimmunotherapy. Regarding dose fractionation, hypofractionated high-dose regimens such as SBRT have been frequently employed. These schedules offer logistical advantages by completing treatment in fewer sessions and are more easily integrated with systemic therapies. Moreover, higher per-fraction doses may be more immunogenic. 98 However, excessively high radiation doses can also lead to immune-related toxicities, including cytokine storms triggered by the abrupt release of large quantities of tumor antigens, and lymphocyte depletion due to collateral damage to normal tissues. As a result, low-dose regional irradiation has been explored as an adjunct strategy. For example, Menon et al. 99 reported that incorporating low-dose radiation improved response rates in non-irradiated lesions, suggesting a potential role in enhancing systemic immune activation. An ongoing registered trial (NCT03800602, ClinicalTrials.gov) is currently investigating the use of low-dose radiotherapy to multiple metastatic sites in conjunction with ICIs. Preliminary findings indicate that multi-site low-dose radiation is feasible and may amplify systemic immune responses. 96 Similarly, in a prospective study by Gkika et al., the immune profiles of 50 patients with early-stage NSCLC who received SBRT were evaluated. SBRT regimens delivering ⩽10 Gy per fraction were associated with significant proliferation of peripheral Ki-67+ PD-1+ CD8+ and CD4+ T cells, together with higher IFN-γ and IL-17A, suggesting that intermediate-dose SBRT may prime tumor-specific T-cell responses and lay a foundation for immunologic synergy. 159 Prospective analyses have also shown that SBRT regimens delivering ⩽10 Gy per fraction can avoid lymphodepletion while simultaneously promoting systemic immune activation. The observed expansion of Ki-67+ CD8+/CD4+ T cells and increased PD-1 expression in peripheral blood support the notion that appropriately fractionated radiotherapy may enhance the immunogenic potential of treatment and facilitate the emergence of abscopal responses. 159
Conversely, not all combinations confer systemic benefit. In the multicenter, randomized phase II CHEERS trial, adding limited-site SBRT (3 × 8 Gy to ⩽3 lesions) to ongoing PD-1/PD-L1 therapy failed to improve PFS or OS, and overall response rates were similar between arms. 145 Most patients had >3 metastases, SBRT was capped at three lesions and delivered before the second/third ICI cycle, and the regimen was explicitly subablative—features that likely constrained out-of-field activity. 145 These data motivate three levers for optimization: maximize safe coverage of dominant disease while preserving TDLNs; use immunogenic yet lymphocyte-sparing dose/fractionation; and align checkpoint blockade within the RT-primed window.
Most clinical trials have adopted either concurrent administration or short-interval sequencing of radiotherapy and ICIs, typically initiating immunotherapy during or within several days to weeks following the completion of radiotherapy. For example, in the aforementioned SWORD study, sintilimab was administered concurrently with SBRT, 30 whereas in the COMIT-1 trial, atezolizumab was initiated immediately after completion of SBRT. 144 Although no prospective trials have directly compared different sequencing strategies in terms of clinical efficacy, retrospective analyses and cohort studies have offered preliminary insights. An immunohistochemical analysis by Van der Woude et al. 65 showed that, in non-irradiated lesions from patients receiving pembrolizumab plus radiotherapy, the tumor microenvironment exhibited favorable immune remodeling (for example, increased infiltration of CD8+ and CD103+ T cells), indicating an immunological abscopal effect reported that immune responses in non-irradiated lesions correlated more strongly with intrinsic features of the TME than with the timing of ICI administration. Conversely, an MD Anderson analysis suggested higher rates of radiographic abscopal responses when PD-1 blockade began after RT completion. 109 Together, these observations support the prevailing practice of starting ICIs during or immediately after RT to capture an “immune window.” While the duration of this window remains incompletely defined, it is generally believed that the weeks immediately following radiotherapy represent a critical period of enhanced neoantigen presentation and T-cell clonal expansion. Administering ICIs during this phase may optimally relieve inhibitory immune checkpoints and promote effective antitumor T-cell responses. 24 A mechanistic study in a murine PDAC model by Hughson et al. exemplified this principle. In that study, IL-12 mRNA was given 24 h after SBRT (6 Gy × 4), with SBRT designed to trigger antigen release and deplete exhausted T-cells, and IL-12 delivered within the ensuing immune window reprogramming T-cell lineages and activating IFN-γ-dependent pathways, thereby clearing non-irradiated liver metastases and establishing durable immune memory. 161 This study provided critical experimental evidence supporting the concept of a “dose–timing–immune activation” axis in the induction of abscopal responses. Additionally, Friedrich et al. developed a biophysical model based on differential equations to simulate the dynamic interactions among tumor cells, T cells, and radiotherapy-induced damage signals. Their findings indicated that the occurrence of abscopal effects is constrained by both radiation dose and ICI timing: low doses may fail to elicit sufficient immunogenicity, whereas excessive doses risk depleting effector T cells. Moreover, ICIs must be administered within the radiotherapy-induced immune activation window to achieve synergy. This model provides a valuable theoretical framework for understanding the mechanisms underlying abscopal effects and for optimizing individualized radioimmunotherapy schedules. 162
Diversification of combination regimens represents a significant advancement in radioimmunotherapy. Beyond the conventional dual approach—RT combined with ICI—increasing efforts are being made to explore triple or even quadruple therapeutic modalities. Representative examples include the SWORD study combining sintilimab, SBRT, and GM-CSF 30 ; a neoadjuvant trial involving tislelizumab, SBRT, and chemotherapy 154 ; and the FFCD 1709 trial, which tested a five-agent regimen consisting of bevacizumab, atezolizumab, SIRT, and chemotherapy. 158 In such regimens, additional agents serve complementary roles: immunostimulatory adjuvants (e.g., GM‑CSF, IMM‑101) increase tumor immunogenicity; anti‑angiogenics (e.g., bevacizumab, anlotinib) normalize vasculature and facilitate immune infiltration95,158; and chemotherapy promotes ICD and antigen release. 154 Across studies, triple regimens have shown signals of improved tumor control with manageable safety. For example, Jiang et al. reported lung cancer with liver metastases treated with local RT, PD‑1 blockade, and anlotinib. Both the irradiated liver lesion and the non‑irradiated primary regressed, consistent with an abscopal response to triple therapy. 95 Similarly, Yang et al. evaluated 8 Gy × 3 SBRT plus intratumoral BCG in a bilateral 4T1 breast tumor model. Intermediate‑dose RT avoided TREX1 upregulation and preserved immunogenicity, while BCG enhanced dendritic‑cell maturation, increased CD8+ T‑cell infiltration, and reduced suppressive cytokines. Together, the regimen induced robust abscopal control and prolonged survival in non‑irradiated lesions, supporting the inclusion of non‑ICI adjuvants in combination strategies. 83 Extending this approach, Caetano et al. added an anti‑MerTK antibody to PD‑1 blockade plus RT, creating a four‑agent strategy. MerTK—a receptor tyrosine kinase on TAMs—promotes apoptotic‑cell clearance and immunosuppression. MerTK blockade increased antigen availability and augmented abscopal responses. 133 These data indicate that co‑targeting multiple nodes of the cancer‑immunity cycle—antigen release/presentation, T‑cell activation, infiltration and effector function, and inhibitory feedback—can synergistically amplify systemic immunity beyond additivity. In another study, Wang et al. combined anaerobic Bifidobacterium (Bi), which accumulates in hypoxic tumor zones, with a monoclonal antibody to trigger complement and antibody‑dependent cellular cytotoxicity (ADCC), forming a quadruple strategy. Integrated with RT and PD‑1 blockade, this platform reversed immunosuppression, enhanced antigen presentation, and increased CD8+ T‑cell infiltration, resulting in significant abscopal control and prolonged survival. 163 Collectively, these findings support a four‑pronged approach—bacterial targeting, antibody activation, RT, and ICI—to convert immunologically “cold” tumors to “hot,” thereby expanding the clinical applicability of abscopal‑based strategies.
As radioimmunotherapy regimens become more complex, systematic toxicity surveillance and management have become central to clinical delivery. When paired with immunotherapy, radiotherapy‑related adverse events—notably pneumonitis and enteritis—can increase in frequency or severity because immune activation may precipitate off‑target autoimmune injury in normal tissues. For lung cancers treated with concurrent chemoradiation plus immunotherapy, respiratory symptoms warrant close, protocolized monitoring. A subset develops severe pneumonitis requiring rapid corticosteroid initiation and escalation pathways. Likewise, SBRT plus dual checkpoint blockade carries overlapping risks of radiation‑induced enteritis and immune‑mediated colitis. In ComIT‑1, grade‑3 treatment‑related events occurred in a subset, yet were generally manageable with standard multidisciplinary care. 144 Across other series, overall adverse‑event rates with combined regimens were not significantly higher than with ICI monotherapy. 30 Collectively, these data indicate that with rational dose selection and careful stratification, a workable efficacy–toxicity balance is achievable; early recognition and tight radiation–medical oncology coordination remain critical to mitigate complications.
Collectively, contemporary clinical data support the antitumor synergy of radiotherapy with immunotherapy. To realize this benefit, key refinements include optimizing dose/fractionation (e.g., SBRT vs conventional; selective low‑dose fields), clarifying ICI sequencing (concurrent vs sequential), and judiciously incorporating immunomodulatory adjuvants or additional agents. At the same time, rigorous toxicity surveillance and proactive management remain essential for patient safety. As datasets mature and intermediate biomarkers are validated, we can delineate patients most likely to benefit and design personalized treatment protocols. Next, we outline emerging predictive biomarkers and individualized approaches to guide clinical decision‑making.
Biomarkers and personalized strategies in radioimmunotherapy
Given that not all patients derive benefit from radioimmunotherapy, identifying reliable biomarkers to predict treatment efficacy—particularly the occurrence of abscopal effects—is of significant clinical importance. Moreover, personalizing treatment on the basis of tumor‑ and patient‑specific features is essential to improve outcomes. This section highlights key candidate biomarkers and individualized approaches that may guide clinical decision-making.
Predictive biomarkers for abscopal responses
Tumor mutational burden (TMB), a surrogate for the quantity of neoantigens, has emerged as a predictive biomarker for ICI efficacy. In RT-ICI settings, high-TMB tumors are theoretically more likely to elicit robust immune responses and abscopal effects owing to enhanced neoantigen release. Sharabi et al. 164 reported a case of advanced neuroendocrine cervical carcinoma with ultra-high TMB that achieved near-complete regression of all lesions after SBRT plus PD-1 inhibition (nivolumab), thereby designating the patient a “hyper-responder.” This case suggests that high TMB can sensitize otherwise ICI‑refractory tumors to RT‑induced neoantigens. Nonetheless, neoantigen quality—rather than quantity alone—remains critical for response.
PD-L1 expression is another commonly used biomarker. High PD-L1 expression typically correlates with better responses to PD-1/PD-L1 blockade. Within the context of radioimmunotherapy, elevated PD-L1 levels may reflect ongoing local immune pressure, which could be further amplified by radiotherapy-induced inflammation and immune checkpoint blockade. In a clinical trial of SBRT combined with atezolizumab in metastatic colorectal cancer, Levy et al. 157 observed that patients with high baseline PD-L1 expression were more likely to experience clinical benefit. Moreover, responders exhibited upregulation of IRF1, indicating a TME with heightened sensitivity to interferon signaling and immunologic activation. Together, PD-L1 and interferon-related transcriptional profiles may serve as indicators of a tumor’s capacity to mount immune responses following radiotherapy.
The abundance and composition of TILs also provide insight into the immune landscape of the tumor. Numerous studies have shown that the presence of activated CD8+ T cells prior to treatment correlates with improved outcomes under immunotherapy. For radioimmunotherapy, tumors with a “hot” immune phenotype—characterized by high TIL density and PD-L1 positivity—are more likely to benefit from radiotherapy, as immune effector cells are already present and ICI targets are pre-existing. These patients may thus be more prone to developing abscopal responses. In a radiographic analysis of immunotherapy-treated RCC, Wong et al. 165 observed that abscopal-like responses and pseudoprogression were often accompanied by T-cell infiltration, clonal expansion, and evidence of antigen spreading. Conversely, in “cold” tumors lacking baseline T-cell infiltration, radiotherapy must first convert the microenvironment into one that can recruit immune cells—a process that may require additional immunostimulatory interventions. Accordingly, baseline TIL levels and transcriptional signatures—including chemokines such as CXCL9/CXCL10—may help predict whether RT-ICI will elicit effective antitumor immune responses. 157
The activity of the cGAS–STING pathway has been proposed as a potential biomarker for predicting abscopal responses. In tumors or associated innate immune cells with intact and responsive STING signaling, radiation-induced DNA damage can be effectively transduced into type I interferon signaling to initiate antitumor immunity. Conversely, deficiencies in this pathway—such as STING gene mutations or loss of downstream signaling components—may significantly impair immune activation post-radiotherapy. Preclinical evidence supports this hypothesis. In a PD-L1 knockout mouse model, Zhao et al. 40 demonstrated that enhancement of abscopal effects by radiotherapy was dependent on cGAS–STING signaling. In human colorectal cancer, Chen et al. 132 reported that interferon-driven upregulation of the immunosuppressive enzyme IDO1 was associated with radioresistance. These findings suggest that patients whose tumors display robust interferon-related gene signatures, indicative of active STING signaling, may benefit from radiotherapy. However, concomitant expression of suppressive mediators such as IDO1 may warrant co‑inhibition (e.g., IDO1 blockade) to overcome resistance. Prospectively, post‑RT quantification of IFN‑β, CXCL10, and other interferon‑response genes could serve as pragmatic surrogates of STING activation and help estimate abscopal probability.
Peripheral blood-based immune profiling also offers a minimally invasive approach to immune monitoring. Biomarkers—including the NLR and circulating T-cell subsets—reflect systemic immune status. Gustafson et al. 166 conducted longitudinal immune profiling in patients with HCC receiving SBRT and found dynamic shifts in lymphocyte subsets that correlated with clinical outcomes. Similarly, Jeon et al. 120 reported that, in breast cancer with bone metastases, SBRT reshaped T‑cell repertoires, including rapid clonal expansion of tumor‑specific clones, consistent with initiation of personalized antitumor immunity. Preclinical data further validate T‑cell receptor (TCR) sequencing as a dynamic readout of systemic immune activation. In a PD‑1-resistant murine lung cancer model, Hu et al. 167 showed that RT combined with NBTXR3 and non‑fucosylated anti‑CTLA‑4/anti‑PD‑1 eradicated primary and metastatic tumors and induced CD8+‑T‑cell activation and clonal expansion, generating durable immune memory. 167 These data suggest that dynamic TCR tracking may help monitor both the onset and durability of abscopal responses. Similarly, Song et al. found that in 4T1 triple‑negative breast cancer, abscopal effects occurred only in radiation‑sensitive tumors, underscoring intrinsic activation thresholds for systemic immunity. When integrated with TCR repertoire analysis, this approach may enable prospective identification of “abscopal responders” based on immunologic potential. 82 Taken together, TCR‑repertoire dynamics plus activation markers (e.g., HLA‑DR+, Ki‑67+ T cells) constitute a pragmatic biomarker suite to gauge immune activation, estimate the likelihood of abscopal responses, and appraise the durability of radioimmunotherapy.
Personalized strategies for radioimmunotherapy
Biomarker-informed approaches offer a promising pathway toward personalized radioimmunotherapy. For patient selection, tumors with an immunologically “hot” phenotype—high TMB, elevated PD‑L1, or abundant TILs—are more likely to benefit from intensified radioimmunotherapy. Such patients may be more prone to abscopal responses and to achieving durable survival outcomes. Conversely, for immunologically “cold” tumors lacking inflammatory signatures, combination strategies should incorporate immune-sensitizing interventions alongside RT-ICI. Examples include STING agonists, tumor vaccines, or metabolic modulators to prime immune recognition, followed by checkpoint blockade to amplify responses. For tumors with intrinsically low STING activity, intratumoral STING agonists or nanoparticle delivery of cGAMP have been shown to enhance abscopal responses. 47 In highly immunosuppressive tumors, personalization can be further guided by the dominant suppressive mechanism indicated by biomarker profiling. For instance, IDO1-overexpressing tumors may benefit from co-administration of IDO1 inhibitors, 132 while tumors with active adenosine signaling may require the addition of CD73 blockade.39,168
In addition to biomarker-guided patient selection, radiation dose and timing can also be tailored to individual immune profiles. For instance, if imaging or biopsy shows low lymphocytic infiltration with myeloid predominance, higher-dose RT or selective low-dose fields directed at adjacent lymphoid structures may be considered to promote lymphocyte recruitment. Conversely, in patients with pre-existing active immune infiltration, a more conservative radiation dose may be preferred to preserve immune effector populations and avoid excessive damage to immunologically engaged tissues. Similarly, dynamic monitoring tools such as ctDNA can provide real-time insights into immunogenic responses. A surge in ctDNA fragments bearing novel mutations shortly after radiotherapy may indicate active neoantigen release, suggesting that immunotherapy should be initiated promptly to capture the ensuing immune activation window. 144 By contrast, absent post‑RT immune activation in peripheral blood may warrant additional interventions—for example, re‑irradiation of another lesion or modification of the immunotherapeutic regimen—to reinvigorate antitumor immunity.
Emerging evidence suggests that distinct metastatic sites may require tailored therapeutic strategies. Liver metastases, in particular, are associated with systemic immune tolerance and may necessitate more intensive interventions to elicit abscopal responses. 90 In patients with hepatic involvement, prioritizing liver‑directed RT may help disrupt local immune suppression. 169 In such cases, dual or even triple immune modulation (e.g., PD-1 plus CTLA-4 blockade, or addition of metabolic-reprogramming agents) may be required to overcome the immunosuppressive hepatic microenvironment. In contrast, pulmonary metastases often reside in an immune-privileged niche rich in resident immune cells. In these patients, radiotherapy combined with a single PD-1 inhibitor may be sufficient to activate systemic antitumor responses. Overall, personalized radioimmunotherapy requires integrated appraisal of tumor-intrinsic features (e.g., molecular biomarkers) and disease-specific factors (e.g., metastatic distribution, prior therapies) to guide combination design. This approach operationalizes precision‑medicine principles in radioimmunotherapy.
With the integration of AI and large-scale data analytics, it is anticipated that predictive models will be developed to estimate individual responses to various radioimmunotherapy regimens. These models can integrate multidimensional inputs—tumor genomics, transcriptomics, radiomics, and clinical features—to inform personalized decisions. An emerging concept is a “radiation-personalized cancer vaccine,” wherein longitudinal TCR-clone tracking pre-/post-treatment infers which neoantigen-specific clones are successfully activated. 28 If mid-treatment analysis reveals clonal expansion of T cells specific to certain antigens, therapies could be intensified along those antigenic pathways. Conversely, a lack of meaningful clonal expansion might prompt adaptive modifications—such as altering the irradiated tumor site or adding immunologic adjuvants. Friedrich et al. further demonstrated, through a mechanistic modeling framework, that the induction of abscopal effects depends on the temporal alignment between radiation-induced antigen release and the immune activation window. This provides a theoretical rationale for individualized scheduling of combined radioimmunotherapy. 162 In addition, AI-based tools may optimize dose distribution to spare immunologically critical organs (e.g., lymph nodes, spleen) and detect early radiographic signs of abscopal responses beyond human perception. Collectively, biomarker‑driven personalization—augmented by AI and systems biology—promises to maximize benefit while minimizing toxicity. Such approaches may expand the clinical relevance of the abscopal effect and enable a broader subset of patients to benefit from systemic immune activation.
Nanotechnology, emerging immunologic targets, and next-generation combination strategies
Beyond conventional immune checkpoint blockade, recent advances in nanotechnology and emerging immunologic targets have introduced new dimensions to radioimmunotherapy. Ongoing studies investigate nanocarrier-mediated targeted delivery of radiosensitizers and immunomodulators to enable precise spatiotemporal control within the TME. Parallel efforts focus on modulating alternative immune pathways—such as metabolic checkpoints and costimulatory receptors—to enhance systemic immune activation and improve the likelihood of abscopal responses. In addition, combining radiotherapy with next-generation immunotherapeutic platforms—including CAR-T cells, oncolytic viruses, and microbiome-based interventions—has shown promising synergistic potential. These innovative strategies represent a new frontier in the rational design of multi-modal regimens aimed at overcoming tumor immune resistance and expanding the clinical applicability of abscopal-based approaches.
Nanotechnology-enabled strategies to potentiate radioimmunotherapy
Nanomedicine has opened new avenues for modulating the TIME, with high spatial and temporal precision in the context of radiotherapy. Recent studies repurpose conventional radiotherapy fiducials as local delivery platforms capable of sustained intratumoral release of immunostimulatory agents. For instance, intratumoral delivery of anti‑CD40 antibodies from fiducial‑like implants enhanced radiotherapy‑induced immune activation and promoted abscopal responses. 170 In lung cancer models, this approach delayed growth of irradiated and non‑irradiated lesions and prolonged survival; comparable activity has been reported in pancreatic and prostate models. Yasmin‑Karim et al. 111 showed that single‑dose radiotherapy combined with nanoparticle CD40 agonism activated CD8+ T‑cell responses and suppressed distant tumor growth—even when irradiation was restricted to sub‑tumoral regions. Zhang et al. developed a gelatinase‑responsive nanoplatform (N@VP) that, when combined with conventional radiotherapy, achieved antitumor efficacy comparable to high‑dose hypofractionation. Mechanistically, this system triggered immunogenic pyroptosis, promoted dendritic‑cell maturation, shifted macrophages toward M1, and generated memory CD8+ T cells, supporting systemic immune activation. Notably, this “in situ vaccine” approach led to significant abscopal tumor control in the absence of immune checkpoint blockade. 108 NBTXR3, a hafnium‑oxide nanoparticle, selectively accumulates in tumors and amplifies intratumoral energy deposition during radiotherapy. Preclinical studies showed that radiotherapy‑activated NBTXR3 increased tumor‑cell death, modified the tumor immunopeptidome to enhance antigen presentation, and increased intratumoral CD8+/CD4+ T‑cell and macrophage infiltration. 53 Intriguingly, similar TCR repertoire reshaping was observed in non-irradiated tumors, suggesting that NBTXR3 amplifies the systemic immune “education” effect of localized radiotherapy. NBTXR3 has demonstrated clinical safety and radiosensitizing efficacy in trials involving soft tissue sarcoma and head and neck cancers, 167 and its combination with immunotherapy may further expand its therapeutic potential. Recent studies have further highlighted the capacity of NBTXR3 to enhance the immunogenicity of proton radiotherapy (PRT). When combined with PD-1 blockade in resistant lung cancer models, this strategy achieved improved local and abscopal tumor control, increased CD8+ T-cell infiltration, suppressed Treg accumulation, and induced broad-spectrum immune memory. 43 Separately, Chen et al. developed an oxygen‑independent radiosensitizer—Fe12‑polyoxometalate (Fe12‑POM)—that triggers Fenton‑like reactions via intermetallic charge transfer upon X‑ray irradiation. This system increased hydroxyl‑radical production and, with anti‑PD‑1 therapy, reprogrammed the microenvironment (e.g., ↑CD80/CD86; ↓FAP+/CD163+), induced IFN‑γ and TNF‑α, and achieved synchronized control across multiple lesions via a strong abscopal effect. 127 Collectively, these data underscore oxygen-independent radiosensitizers as promising enhancers of immunotherapy efficacy in hypoxic solid tumors.
Another major class of nanotechnology-based strategies involves the targeted delivery of immune activators to enhance the immunogenicity of radiotherapy. Wang et al. developed a multifunctional nanoparticle system (DMPtNPS@cGAMP) co-loaded with the STING agonist cGAMP and platinum-based components. This platform enabled efficient intratumoral delivery of cGAMP to activate STING signaling and alleviate tumor hypoxia via platinum-induced ROS; when combined with radiotherapy in a rectal cancer model, it significantly amplified type I interferon signaling, promoted CD8+ T-cell infiltration, and suppressed distant tumor growth. 46 Similarly, Li et al. reported a near-infrared IIb-emitting nanoplatform capable of inducing immunogenic photodynamic damage and releasing tumor antigens within irradiated lesions. This platform synergized with radiotherapy to induce systemic antitumor immunity and control distant metastases in a multifocal tumor model. 52 CAF targeting has also emerged as a promising adjunct in radioimmunotherapy. Zhou et al. developed a FAPα-responsive photosensitizer probe (FMP) that selectively ablates CAFs and induces ICD. When combined with PD-L1 blockade, this approach triggered robust abscopal responses and durable systemic immune memory. 55 Thermosensitive liposomes carrying immunomodulatory payloads represent another precision delivery modality. Regenold et al. engineered liposomes encapsulating IL-12 that released their contents upon heating following radiotherapy. In a pancreatic tumor model, this system enhanced M1 macrophage polarization and CTL infiltration, resulting in complete tumor regression and abscopal effects. 77 Gao et al. 128 designed a hydrogel‑based CCL21‑DCs@gel platform that sustained chemokine release, significantly promoted immune‑cell infiltration after radiotherapy, and triggered abscopal responses in melanoma models—illustrating the potential of an in situ vaccination strategy based on local post‑radiotherapy immunostimulation. 128 Recently, Au et al. 91 introduced a redox-responsive, injectable tumor immune niche (TIN) nanofiber platform that released α-CTLA-4, IL-2-Fc fusion proteins, and poly(I:C) under radiotherapy-induced hypoxic and reductive conditions, thereby significantly amplifying local and systemic antitumor immunity in melanoma and colorectal cancer models and leading to complete responses and long-term immune memory in a subset of animals. Zhang et al. proposed a tri-modal nanovaccine strategy involving IL-2 and iron oxide/polylysine/CpG-based nanoparticles (PICs). When injected into irradiated tumors, this platform enhanced antigen presentation, Th1/M1 polarization, and systemic CD8+ T-cell activation. In a bilateral tumor model, the combination induced potent abscopal responses and long-lasting immune memory, providing a translatable tool for in situ vaccination enhancement. 171 In preclinical models, an injectable oxygen microparticle system locally relieved hypoxia in irradiated tumors and, with radiotherapy, significantly increased CD8+ T-cell infiltration and systemic antitumor immunity, thereby inducing pronounced abscopal effects and highlighting oxygen modulation as a promising lever to amplify in situ vaccine-like immune responses. 50 Collectively, these studies demonstrate that nanocarriers can function as “guided missiles” to precisely deliver immunologic payloads into the TME. When timed with radiotherapy, these platforms enable spatiotemporal control of immune activation, transforming localized treatment into systemic therapeutic outcomes. A summary of representative nanotechnology-based platforms and their roles in enhancing radiotherapy-induced abscopal effects is presented in Table 3.
Novel nano-immune technologies enhancing radiotherapy-induced abscopal effects.

BNCT, boron neutron capture therapy; B-NIT, Boron Neutron Immunotherapy; CAF, cancer-associated fibroblast; Cu, copper; DC, dendritic cells; DSF, disulfiram; ECM, extracellular matrix; HMGB, high-mobility group box 1; ICD, immunogenic cell death; ICI, immune checkpoint inhibitors; IDO1i, IDO1 inhibitors; MG, microbial metabolite; NK, natural killer; ROS, reactive oxygen species; RT, radiotherapy; SBRT, stereotactic body radiotherapy; TAA, tumor-associated antigens; TIME, tumor immune microenvironment; TIN, tumor immune niche; TME, tumor microenvironment. CDRT, conventional-dose radiotherapy; DAMP, damage-associated molecular pattern(s); ER, endoplasmic reticulum; TEM, T effector memory (cells).
Beyond acting as carriers or radioenhancers, selected nanomaterials also exhibit intrinsic immunomodulatory activity, which can be strategically integrated into radioimmunotherapy. For instance, Shen et al. 169 designed a metabolic homeostasis-regulating nanoparticle (Metabolic NP) that co-releases mannose and levamisole to concurrently inhibit glycolysis and mitochondrial respiration in tumor cells, thereby weakening cancer-cell stress adaptation and reprogramming metabolic competition within the TME. In murine models, Metabolic NPs combined with radiotherapy significantly reduced M2-like macrophages and Tregs, enhanced effector T-cell activity, and elicited abscopal responses. 169 Similarly, Pang et al. developed a bioactive polysaccharide-based nanoparticle (ANP) that directly engages DCs, promoting maturation and antigen presentation. ANP treatment increased intratumoral CD4+/Treg and CD8+/Treg ratios, enhanced systemic antitumor immunity and immune memory, and, when combined with radiotherapy, suppressed the growth of non-irradiated metastases. 129 More recently, Li et al. 172 reported the hybrid nanoplatform THUNDER, which synchronizes antigen release from hypoxic and normoxic regions and co-activates the DC/CD8+ T-cell axis, thereby inducing robust abscopal responses and long-term immune memory in a breast cancer model. Combined with PD-L1 blockade, the platform further augmented systemic immunity, illustrating a tri-modality RT-ICI–nanotechnology strategy. 172 Collectively, these studies indicate that nanomaterials can act as potent immunologic modulators without added adjuvants. Such approaches offer alternatives for patients’ intolerant of conventional immunostimulants and may expand the therapeutic window of radioimmunotherapy.
Emerging immune targets and innovative combination strategies
In addition to classical immune checkpoints such as PD-1 and CTLA-4, a growing number of emerging immunoregulatory molecules have shown potential as synergistic partners for radiotherapy. One well-characterized example is the CD73–adenosine signaling axis. CD73 enzymatically converts extracellular ATP into adenosine, a potent immunosuppressive metabolite that inhibits the function of various immune cell subsets. Multiple preclinical studies have shown that combining CD73 inhibitors with radiotherapy significantly reduces immunosuppressive adenosine in the TME and increases both the abundance and functionality of tumor-specific T cells.39,168 An et al. 39 further reported that CD73 blockade potentiated radiation‑induced cGAS–STING signaling, increased dendritic‑cell and CD8+ T‑cell infiltration, and augmented abscopal responses in murine models. These findings position CD73 as a promising co-target in the context of radioimmunotherapy. Currently, both anti-CD73 monoclonal antibodies and small-molecule inhibitors are in clinical evaluation, and integrating them with radiotherapy or ICI-based regimens is a promising way to broaden immune-enhancing combination strategies.
Tumor metabolism-associated targets have garnered increasing attention as adjuncts in radioimmunotherapy. SHP2, a protein tyrosine phosphatase expressed in both tumor and immune cells, has been implicated as a key regulator of immunosuppressive signaling within the TME. In a PD-1-resistant NSCLC mouse model, Chen et al. 79 showed that SHP2 inhibition (SHP099), combined with radiotherapy and anti-PD-L1, significantly improved primary and distant tumor control and survival, and enhanced antitumor immunity by increasing CD8+ T-cell infiltration, reducing Tregs, and promoting M1 macrophage polarization. Several other immunometabolic molecules—including IDO1, 132 CD36, 123 and VEGFR2 122 —have similarly been shown to augment radiotherapy-induced systemic immune responses when pharmacologically inhibited. Epigenetic regulators have also emerged as promising radiosensitization targets. BRD4, a histone reader protein, contributes to immune evasion by upregulating HIF-1α, PD-L1, and chemotactic signals for immunosuppressive cells. In a murine breast cancer model, Kim et al. 119 found that BRD4 inhibition enhanced CD8+ T-cell infiltration and delayed tumor progression when combined with radiotherapy. In a rat breast cancer model, the HDAC inhibitor valproic acid induced abscopal responses by reprogramming the immune landscape of non-irradiated (distant) tumors—enhancing post-radiotherapy CD8+ T-cell infiltration, promoting M1 macrophage polarization, reducing Tregs, and upregulating IL-12 and IL-1β—thereby increasing their immunogenicity. 173 These findings underscore the potential of HDAC inhibitors as immune-sensitizing agents in radiotherapy. Metformin, a metabolic drug with well-characterized immune effects, has also been shown to induce abscopal suppression of pulmonary metastases in a rectal cancer model when combined with radiotherapy. Mechanistically, this effect was mediated by increased activity of IFN-γ-producing T cells and NK cells, along with reversal of T-cell exhaustion. 136 Similarly, ATRA, a differentiation agent targeting metabolic pathways, has been shown to reprogram the TME by inducing inflammatory macrophages and enhancing CD8+ and CD4+ T-cell responses, and—in murine models—to elicit robust abscopal responses when combined with radiotherapy via an IFN-γ-dependent inflammatory loop. 118 Together, these studies highlight that metabolic and epigenetic modulation can serve as effective adjuncts to radioimmunotherapy by reconditioning the tumor immune landscape and enhancing systemic immune responsiveness.
While CAR-T therapy has demonstrated remarkable success in hematologic malignancies, its efficacy in solid tumors remains limited due to poor infiltration, immunosuppressive TME, and antigen heterogeneity. However, emerging evidence suggests that radiotherapy can act as a “priming” modality to facilitate CAR-T-cell function. Wang et al. 57 reported that radiation upregulated B7-H3 and death receptors (e.g., Fas) on tumor cells, sensitizing tumors to B7-H3-targeted CAR-T therapy. 57 In a bilateral tumor model, irradiating one tumor followed by systemic CAR-T infusion led not only to regression of the irradiated lesion but also to enhanced CAR-T activity in the non-irradiated site—demonstrating a CAR-T-mediated abscopal effect in solid tumors. Similarly, in a pancreatic cancer model, radiotherapy plus CLDN18.2–CAR-T increased tumor-cell CCL2, recruiting CAR-T cells and improving metastatic control. 113 Related trimodality approaches are in early clinical testing; for example, in the SACTION01 trial, Zhao et al. 154 reported 76% major pathological response in early-stage NSCLC treated with neoadjuvant SBRT (24 Gy in three fractions) plus anti-PD-1 tislelizumab and chemotherapy, underscoring the synergy of radiotherapy–immunotherapy–chemotherapy in resectable lung cancer. Additionally, Amit et al. 69 found that PRT, via Bragg-peak properties, more effectively upregulated TAAs (e.g., mesothelin) while limiting off-target tissue damage in pancreatic tumors. Combined with mesothelin-targeted CAR-T cells, proton radiation outperformed photon radiation in enhancing antitumor immune responses. 69 Collectively, these studies support radiation preconditioning (“pre-adaptation”) to enhance infiltration, activation, and persistence of adoptive cellular therapies (CAR-T/TCR-T) in solid tumors. 57 Ongoing trials are evaluating low-dose radiotherapy preconditioning before CAR-T infusion in refractory solid tumors to improve persistence and intratumoral distribution.
Microbiome‑directed interventions are increasingly recognized as tractable levers to enhance systemic antitumor immunity. The gut microbiota shapes responsiveness to immune checkpoint blockade, and radiotherapy can, in turn, alter microbial composition. Select probiotic strains generate immunoactive metabolites that promote host antitumor immunity. MG—a gut microbiota-derived metabolite—has been identified as a radiosensitizer and immunomodulator. In syngeneic rectal cancer, MG augmented responses to radiotherapy and anti-PD‑1 by precipitating endoplasmic reticulum stress, elevating ROS, and engaging the cGAS–STING pathway. 19 MG improved control of both irradiated and contralateral non‑irradiated lesions, supporting its development as a microbiome‑linked adjunct to radioimmunotherapy. Translationally, strategies that raise intratumoral MG—dietary modulation or targeted probiotic supplementation—warrant prospective evaluation to potentiate RT‑induced immunity; safety windows (e.g., glycation‑related toxicities) require definition prior to clinical testing. In parallel, Wang et al. 163 engineered an anaerobic bacterium–antibody (Bi + mAb) platform that preferentially targets hypoxic tumor niches and activates complement/ADCC; when integrated with radiotherapy and PD‑1 blockade in bilateral 4T1 and CT26 models, it increased CD8+ T‑cell infiltration and antigen presentation, suppressed MDSCs, and strengthened abscopal control—consistent with microbiome‑driven “cold‑to‑hot” conversion. Engineered bacteria have also been explored as delivery vectors for radiosensitizing agents. Zhang et al. showed that engineered bacteriophages and Escherichia coli delivering CD40 ligand or AIEgen-based radiosensitizers enhanced local and systemic immunity, thereby amplifying radiotherapy abscopal effects.29,72 Similarly, the VNPNKase system released nattokinase systemically, remodeling the TME and improving radioimmunotherapy synergy, comparable to the “in situ immune activation” seen with phage-based platforms. 71 Clinically, in pancreatic cancer—an archetypal “cold” tumor—SBRT combined with heat‑inactivated Mycobacterium obuense (IMM‑101) increased peripheral T‑ and NK‑cell activation and correlated with improved PFS. 112 Collectively, these microbiome-associated strategies bridge local tumor-directed therapies with systemic metabolic and immune regulation, representing a multidisciplinary evolution in the design of radioimmunotherapeutic interventions.
Oncolytic virotherapy and mRNA-based immunotherapies represent emerging modalities that can synergize with radiotherapy to enhance systemic antitumor immunity. BO‑112, a nanoplexed synthetic double‑stranded RNA (poly‑I:C) that activates TLR3/MDA5 signaling, exemplifies this class. Administered intratumorally, BO‑112 can synergize with radiotherapy by enhancing dendritic‑cell cross‑priming and CD8+ T‑cell activation, thereby potentiating immune‑mediated tumor control. 124 IL‑12 mRNA nanoparticles offer localized cytokine delivery that can reverse T‑cell dysfunction and restore cytotoxic activity within irradiated tumors. In preclinical models, radiotherapy combined with an OX40 agonist and CpG (a “triplet vaccine”) elicited robust systemic immunity, and addition of PD‑1 blockade produced durable tumor eradication. 58 As mRNA platforms mature, programmable strategies—such as in situ cytokine factories or neoantigen‑encoding RNA—are being integrated with radiotherapy to improve antigen recognition. 161 Co‑delivery of cytokines with multifunctional nanoparticles can reinforce abscopal responses; for example, intratumoral IL‑2 plus PIC nanoparticles synergized with radiotherapy to remodel distant, non‑irradiated lesions. 171
In summary, emerging nanotechnologies and next‑generation immune targets have broadened the therapeutic repertoire of radioimmunotherapy. Clinically, the traditional “RT + ICI” model is progressing toward triplet designs (RT + X + Y), in which X/Y include nanocarriers, metabolic inhibitors, costimulatory agonists, or cellular therapies. This multidimensional strategy aims to tackle persistent barriers—leveraging nanotechnology to address heterogeneity and delivery, using novel targets to dismantle dominant immunosuppressive circuits, and converting “cold” tumors to “hot” through multimodal synergy. However, clinical deployment requires careful dose–sequence optimization and vigilant toxicity management, because regimen complexity introduces both logistical and biological risks. As enabling technologies mature and tumor immunology advances, clinically vetted multi‑agent regimens are poised to enter routine practice. Within this framework, radiotherapy functions as an immunologic trigger rather than a purely local cytotoxin, synergizing with programmable platforms to maximize durable tumor control and, in selected settings, cure.
Future perspectives
Radioimmunotherapy is rapidly transitioning from proof‑of‑concept to clinical application and now stands at a pivotal juncture in its evolution as a frontline anticancer strategy. Looking ahead, priorities include refining and personalizing RT; integrating cross‑disciplinary innovations (e.g., AI); bridging preclinical–clinical translation; and developing strategies for resistant, immunologically “cold” tumors.
First, precision and spatiotemporal optimization of RT delivery will continue to advance. Advanced modalities—FLASH, proton/heavy‑ion, spatially fractionated RT (microbeam/GRID), and image‑guided adaptive RT—aim to maximize tumor control while sparing immune function. For example, FLASH uses ultrahigh dose rates to reduce normal‑tissue toxicity and inflammation, preserving lymphocytes and improving compatibility with immunotherapy. 27 Spatially fractionated modalities, including GRID or microbeam techniques, generate alternating high- and low-dose regions within the tumor. By integrating ablative “peaks” with permissive “valleys” within a single target, SFRT preserves intratumoral APC/lymphocyte niches while amplifying cGAS–STING/type-I IFN signaling and chemokine-guided T-cell recruitment; early lattice and SBRT-PATHY series, together with partial-volume or minibeam proton/carbon reports, describe non-targeted regressions with low toxicity, warranting prospective immuno-oncology trials to standardize geometry and sequencing.103,105,174,175 Trappetti et al. 49 demonstrated vaccine-like priming with multibeam micro-irradiation in melanoma. SBRT-PATHY—which time-synchronizes hypoxia-targeted irradiation while sparing immune-infiltrated regions—has been associated with higher abscopal rates in early clinical series. 176 Collectively, these innovations mark a paradigm shift: RT aims not only to eradicate tumor cells but to orchestrate immune‑mediated elimination. Accordingly, dose, target volume, fractionation, and timing become programmable variables. With immunologically informed optimization, RT can both debulk disease and prime systemic antitumor immunity.
Second, AI and machine learning are expected to play an increasingly important role in the personalized design of radioimmunotherapy. AI can integrate clinical, imaging, and molecular datasets to uncover predictive patterns that inform treatment. For example, models can estimate abscopal‑response probability after RT + ICI using baseline radiomics and tumor immune‑gene expression. Such tools can aid regimen selection and set realistic expectations. ML may also enable real‑time adaptation of RT plans. By monitoring immunologic biomarkers during treatment, algorithms could dynamically adjust dose or fields to maximize immune activation. Early efforts exist: Ojlert et al. used pre‑/post‑RT peripheral TCR sequencing from NSCLC patients in ML models to capture dynamic shifts in TCR diversity. Expansion or contraction of T‑cell clones correlated with efficacy. 28 Such AI‑assisted systems could be embedded into clinical decision workflows. For instance, expanding TCR clones might support continuation, whereas absence expansion could prompt timely modification (e.g., adding immunotherapies). AI also holds promise for composite‑biomarker discovery. By jointly analyzing PD‑L1, TMB, TIL density, and other features, AI may enable integrated immune scores with improved predictive accuracy. As radioimmunotherapy datasets grow, AI will better decipher complex biology and translate it into actionable guidance.
Third, overcoming the translational barriers between preclinical discovery and clinical implementation remains a central challenge. Although enhanced abscopal responses are frequent in preclinical models, clinical occurrences remain uncommon. This discrepancy reflects several factors. The human tumor–immune microenvironment is inherently more complex than in murine models, and host immunity is shaped by age, comorbid disease, chronic infection, and other variables. Furthermore, regimens effective in mice may require higher intensity or more frequent dosing in humans to achieve comparable efficacy—often breaching safety limits and becoming clinically infeasible. To address these limitations, adaptive trial designs and innovative dose‑delivery strategies are needed to balance efficacy and tolerability. Additionally, robust evaluation of abscopal responses remains methodologically challenging. Traditional criteria such as RECIST often miss non‑conventional patterns, including abscopal regression and pseudoprogression. Incorporating immune‑related radiographic endpoints, ctDNA dynamics, and other immunologic biomarkers as co‑primary or exploratory endpoints will be critical to accurately assess therapeutic benefit and to guide future regimen refinement. 137 There is increasing interest in centralized registries dedicated to abscopal responses. Such registries could aggregate clinical cases, enable pattern recognition, and clarify the conditions under which these rare yet promising responses are most likely to occur.
Finally, addressing immunologically “cold” tumors and treatment‑resistant disease will require intensified, innovative strategies. These tumors often respond poorly to ICIs or conventional radioimmunotherapy and therefore demand more aggressive “immune‑heating” approaches. Advances in nanotechnology, novel immunologic adjuvants, and multi‑target combinations are being developed to address this challenge. For instance, traditionally refractory malignancies such as pancreatic and colorectal cancer may benefit from integrated multimodal regimens. These may include high‑dose SBRT (e.g., CyberKnife), intratumoral STING agonists, dual checkpoint blockade (PD‑1/PD‑L1 plus CTLA‑4), and microbiome modulation. This multimodal approach may ultimately enable some cold tumors to transition into more manageable, chronic conditions. Emerging clinical observations in MSS colorectal cancer with liver metastases support the feasibility of this strategy.158,177 Importantly, implementing such complex regimens places substantial demands on healthcare systems. It requires seamless collaboration among multidisciplinary teams—including radiation oncologists, medical oncologists, interventional radiologists, and immunology laboratories—and substantial patient engagement and tolerance. Nonetheless, if these strategies deliver meaningful survival gains, patients and clinicians are likely to accept the trade‑offs inherent to such intensive yet promising therapies.
Conclusion
The abscopal effect—a rare but compelling regression of distant lesions following local radiotherapy—holds substantial mechanistic and translational promise. In treatment‑refractory solid tumors (e.g., colorectal cancer with liver metastases)—where standard options often fail—radioimmunotherapy has emerged as a promising strategy to improve outcomes. This review outlines the molecular underpinnings of RT‑induced immune activation, including DNA‑damage-triggered cGAS–STING activation, antigen release via ICD, and remodeling of immune cell populations within the TME. A mechanistic understanding of how RT simultaneously “presses the accelerator” of immune activation and “applies the brakes” via immunosuppressive circuits is essential for the rational design of effective combination strategies.
The synergy between radiotherapy and ICIs is supported by multiple preclinical models and clinical trials. PD‑1/PD‑L1 blockade reverses T‑cell exhaustion, whereas CTLA‑4 inhibition augments antitumor T‑cell priming. Together with radiation‑induced neoantigen release, these mechanisms form a complementary framework amplifying systemic immunity. Treatment sequencing and radiation dose strongly influence efficacy, underscoring the need for patient‑tailored regimens. Preclinical studies delineate the contributions of Tregs, MDSCs, DCs, macrophages, and NK cells to abscopal control. These mechanistic insights inform strategies to enhance radioimmunotherapy. These discoveries are already driving clinical innovation—triplet/quadruplet regimens, integration of targets (CD73, IDO1, STING), and adjuvants (GM‑CSF, vaccines, metabolic modulators). Collectively, these efforts mark a shift toward rational, mechanism‑based designs to unlock the full potential of RT-immunotherapy. Taken together, these insights form the basis of a translational paradigm shift—from viewing radiotherapy as a local cytotoxic modality to recognizing it as a system-wide immune enabler. This evolving view opens new frontiers for designing precision combination therapies, optimizing timing, and personalizing immune engagement in a tumor-specific manner.
Current clinical evidence indicates that radioimmunotherapy can deliver exceptional benefit in a subset of patients—from dramatic tumor regression to prolonged survival—even after failure of standard therapies. However, many patients derive limited or no benefit, underscoring the need to acknowledge persistent unmet needs alongside successes. Key challenges include: (i) deploying predictive and dynamic biomarkers to identify likely responders and guide combination selection; (ii) overcoming the immunologic barriers of “cold” tumors to convert non‑responders; and (iii) tightly controlling toxicity so multi‑modal regimens maximize benefit without compromising safety. Addressing these issues will require sustained mechanistic studies, biomarker discovery, and rigorously designed, cross‑disciplinary clinical trials. Ultimately, progress on these fronts is essential to unlock the full potential of radioimmunotherapy and extend its benefits to more patients.
In summary, mechanistic insights into radiation‑induced abscopal immunity have deepened our understanding of tumor immunology and catalyzed new therapeutic approaches for solid tumors. The integration of radiotherapy and immunotherapy has moved from complementary to synergistic and is increasingly viewed as a prospective cornerstone of oncologic care. With continued advances in technology and conceptual frameworks, radioimmunotherapy should further improve outcomes across malignancies, potentially extending survival and enabling durable responses. Realizing this potential will require sustained basic–clinical collaboration and rigorous, evidence‑driven evaluation of emerging hypotheses and strategies. Such interdisciplinary rigor is essential to render the abscopal effect a consistent, clinically translatable modality that benefits a broader patient population.
Footnotes
Acknowledgements
The authors thank their colleagues for their general support and helpful discussions during the preparation of this manuscript.
Declarations
Use of generative AI
The authors affirm that no generative AI tools (e.g., ChatGPT or similar) were used in any part of the manuscript preparation, including writing, data generation, figure creation, or reference management.